

Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review
- PMID: 29228169
- PMCID: PMC7729208
- DOI: 10.1093/cvr/cvx226
Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review
Abstract
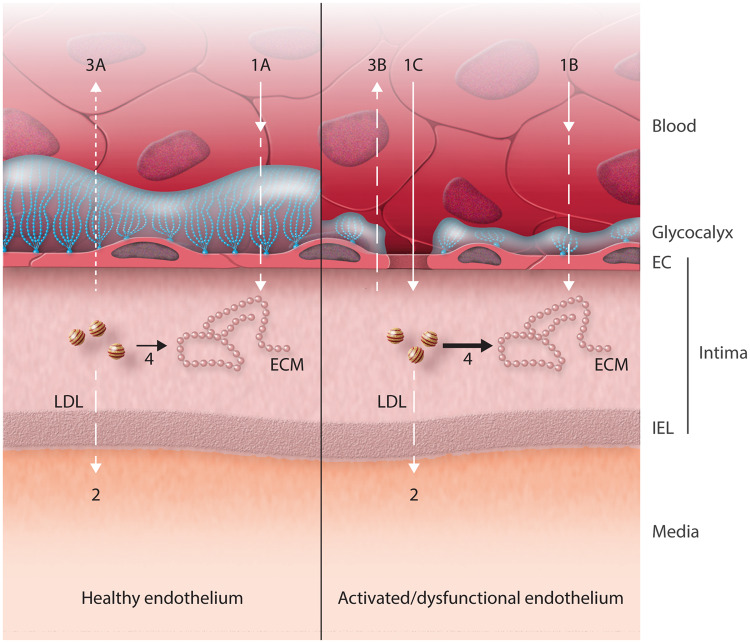
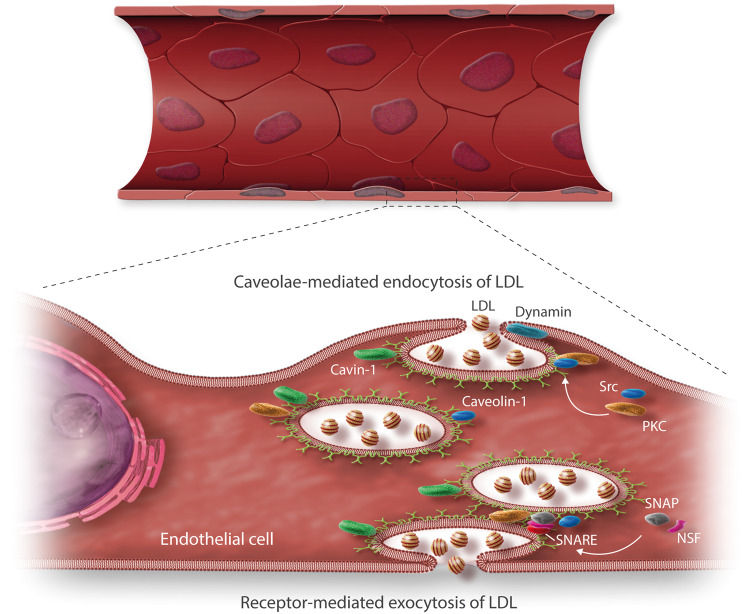
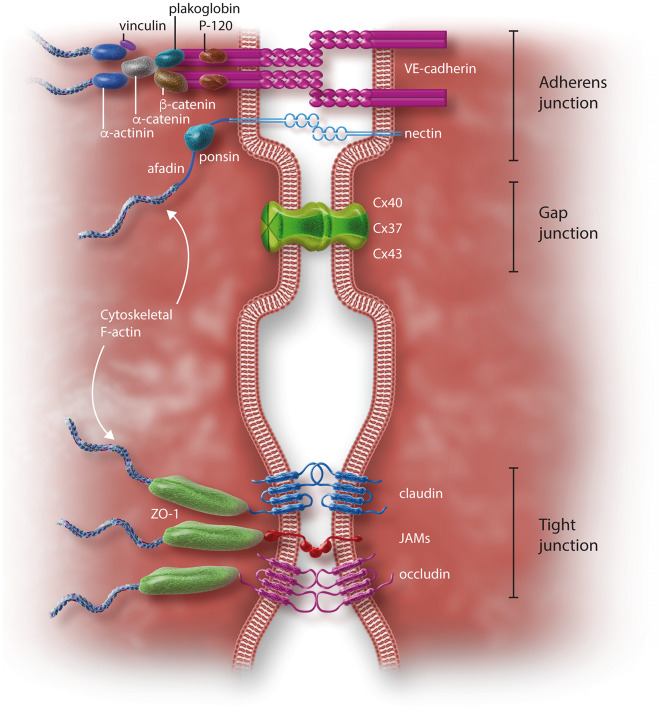
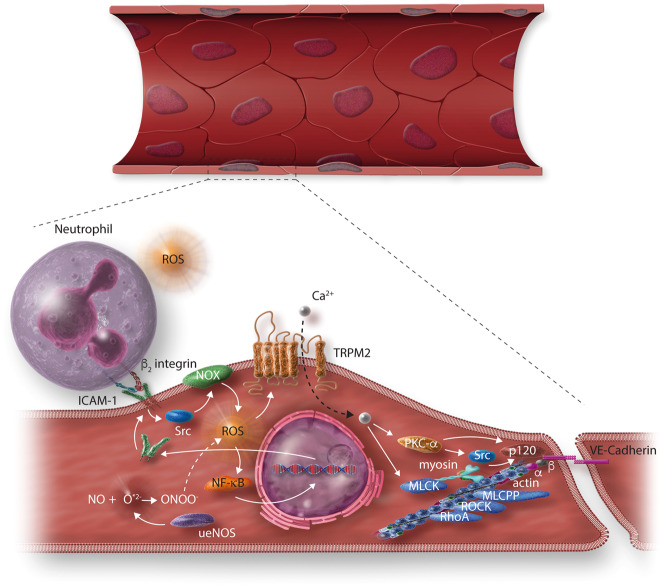
Early atherosclerosis features functional and structural changes in the endothelial barrier function that affect the traffic of molecules and solutes between the vessel lumen and the vascular wall. Such changes are mechanistically related to the development of atherosclerosis. Proatherogenic stimuli and cardiovascular risk factors, such as dyslipidaemias, diabetes, obesity, and smoking, all increase endothelial permeability sharing a common signalling denominator: an imbalance in the production/disposal of reactive oxygen species (ROS), broadly termed oxidative stress. Mostly as a consequence of the activation of enzymatic systems leading to ROS overproduction, proatherogenic factors lead to a pro-inflammatory status that translates in changes in gene expression and functional rearrangements, including changes in the transendothelial transport of molecules, leading to the deposition of low-density lipoproteins (LDL) and the subsequent infiltration of circulating leucocytes in the intima. In this review, we focus on such early changes in atherogenesis and on the concept that proatherogenic stimuli and risk factors for cardiovascular disease, by altering the endothelial barrier properties, co-ordinately trigger the accumulation of LDL in the intima and ultimately plaque formation.
Keywords: Atherosclerosis; Cardiovascular risk factors; Endothelium; Vascular permeability.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2017. For permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Oxidative stress in atherosclerosis: the role of microRNAs in arterial remodeling.Free Radic Biol Med. 2013 Sep;64:69-77. doi: 10.1016/j.freeradbiomed.2013.06.025. Epub 2013 Jun 21. Free Radic Biol Med. 2013. PMID: 23797034 Review.
-
Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis.Arterioscler Thromb Vasc Biol. 2014 Mar;34(3):509-15. doi: 10.1161/ATVBAHA.113.300156. Arterioscler Thromb Vasc Biol. 2014. PMID: 24554606 Review.
-
Highlight on Endothelial Activation and Beyond.Arterioscler Thromb Vasc Biol. 2018 Dec;38(12):e198-e201. doi: 10.1161/ATVBAHA.118.312054. Arterioscler Thromb Vasc Biol. 2018. PMID: 30571176 Review. No abstract available.
-
[Oxidative stress and atherosclerosis].Orv Hetil. 2015 Jul 12;156(28):1115-9. doi: 10.1556/650.2015.30201. Orv Hetil. 2015. PMID: 26149503 Review. Hungarian.
-
Patient-specific computational modeling of subendothelial LDL accumulation in a stenosed right coronary artery: effect of hemodynamic and biological factors.Am J Physiol Heart Circ Physiol. 2013 Jun 1;304(11):H1455-70. doi: 10.1152/ajpheart.00539.2012. Epub 2013 Mar 15. Am J Physiol Heart Circ Physiol. 2013. PMID: 23504178
Cited by
-
Preventive Therapies in Peripheral Arterial Disease.Biomedicines. 2023 Nov 27;11(12):3157. doi: 10.3390/biomedicines11123157. Biomedicines. 2023. PMID: 38137379 Free PMC article. Review.
-
Sinner or Saint?: Nck Adaptor Proteins in Vascular Biology.Front Cell Dev Biol. 2021 May 26;9:688388. doi: 10.3389/fcell.2021.688388. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34124074 Free PMC article. Review.
-
ITGB2 is a central hub-gene associated with inflammation and early fibro-atheroma development in a swine model of atherosclerosis.Atheroscler Plus. 2023 Nov 15;54:30-41. doi: 10.1016/j.athplu.2023.11.001. eCollection 2023 Dec. Atheroscler Plus. 2023. PMID: 38116576 Free PMC article.
-
Growth hormone-releasing hormone antagonists protect against hydrochloric acid-induced endothelial injury in vitro.Environ Toxicol Pharmacol. 2023 Apr;99:104113. doi: 10.1016/j.etap.2023.104113. Epub 2023 Mar 20. Environ Toxicol Pharmacol. 2023. PMID: 36940786 Free PMC article.
-
Effects of PCSK9 inhibitors on coronary microcirculation, inflammation and cardiac function in patients with CHD after PCI: a protocol for systematic review and meta-analysis.BMJ Open. 2023 Sep 18;13(9):e074067. doi: 10.1136/bmjopen-2023-074067. BMJ Open. 2023. PMID: 37723117 Free PMC article.
References
-
- De Caterina R, Libby P. Endothelial Dysfunctions in Vascular Disease. Oxford, UK: Blackwell Publishing, 2007.
-
- Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, Boren J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002;417:750–754. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
