

The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions
- PMID: 29227476
- PMCID: PMC5907936
- DOI: 10.1038/nm.4439
The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions
Erratum in
-
Erratum: The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions.Nat Med. 2018 Apr 10;24(4):526. doi: 10.1038/nm0418-526b. Nat Med. 2018. PMID: 29634680
-
Publisher Correction: The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions.Nat Med. 2019 Mar;25(3):530. doi: 10.1038/s41591-019-0369-7. Nat Med. 2019. PMID: 30705421 Free PMC article.
Abstract
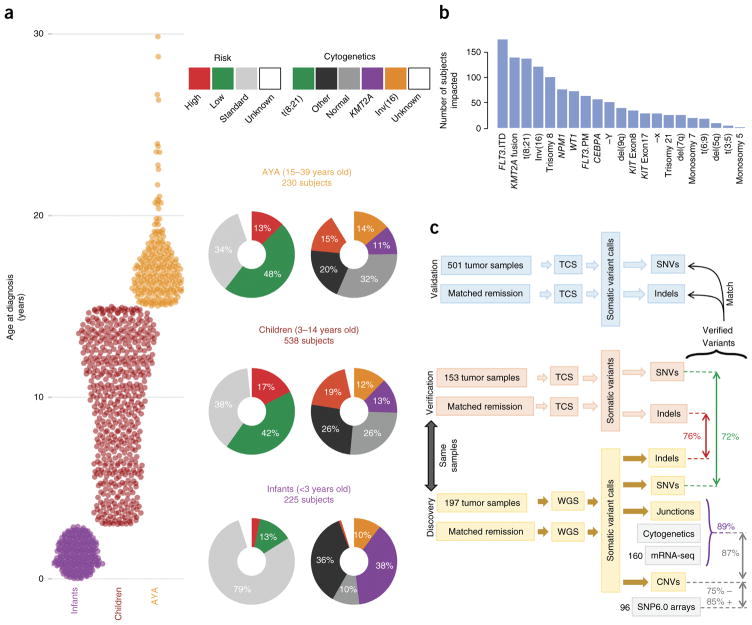
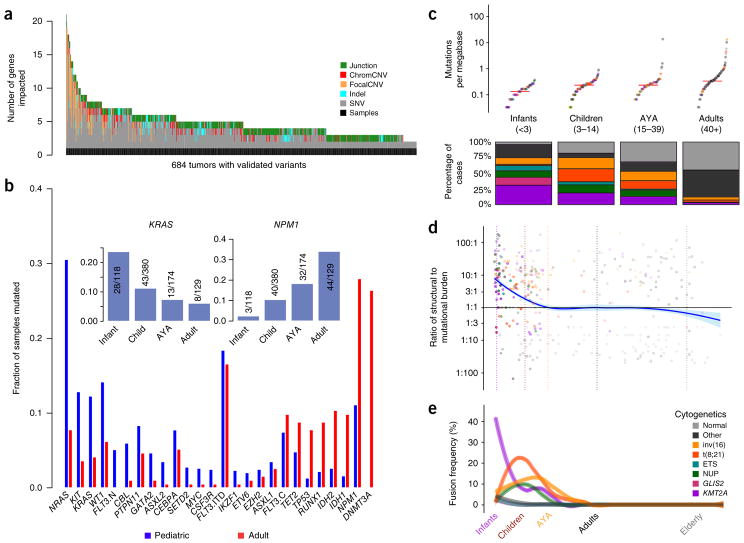
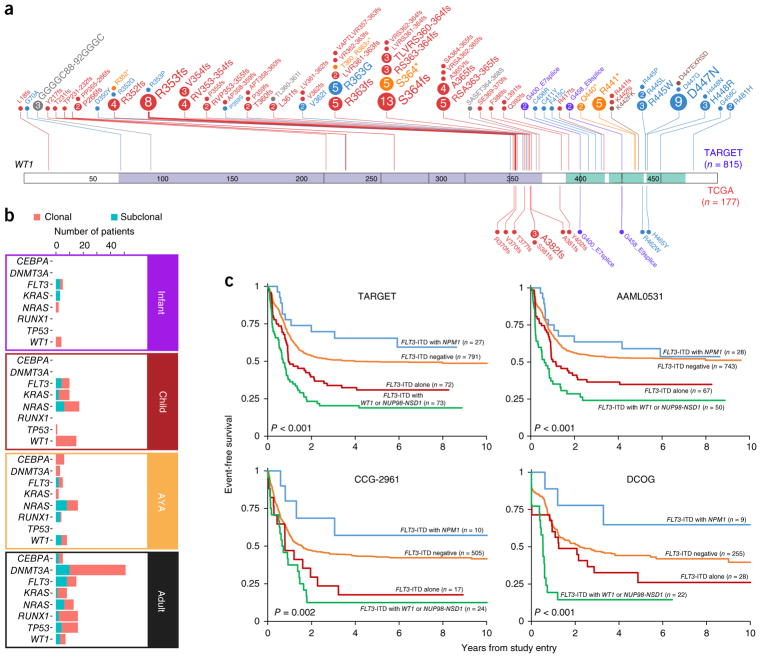
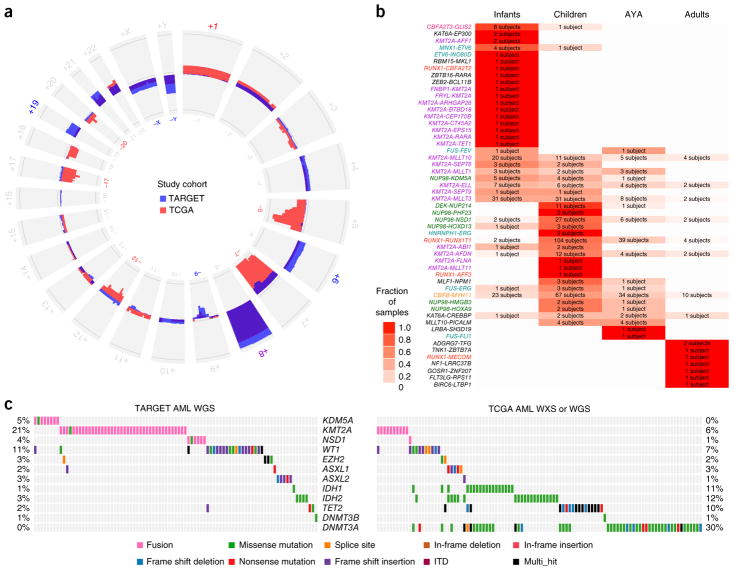
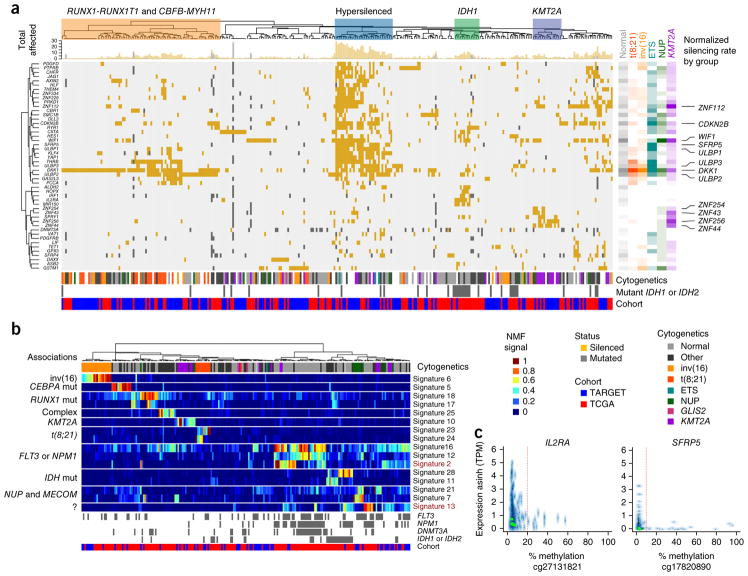
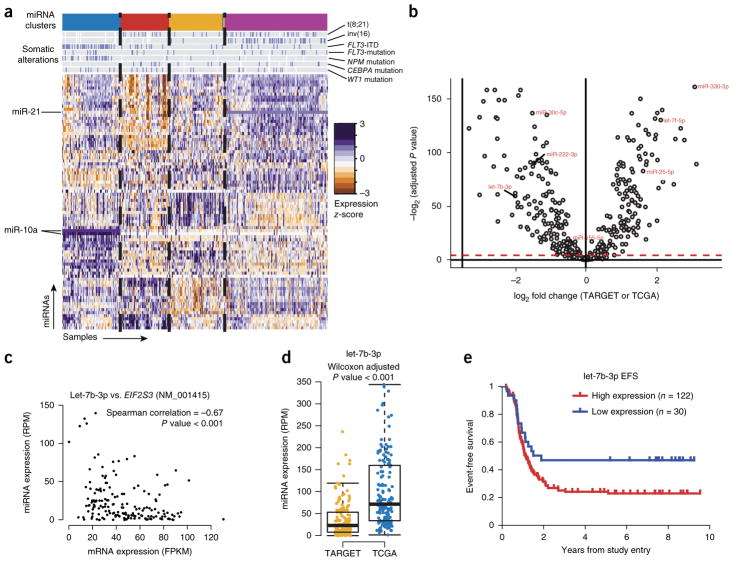
We present the molecular landscape of pediatric acute myeloid leukemia (AML) and characterize nearly 1,000 participants in Children's Oncology Group (COG) AML trials. The COG-National Cancer Institute (NCI) TARGET AML initiative assessed cases by whole-genome, targeted DNA, mRNA and microRNA sequencing and CpG methylation profiling. Validated DNA variants corresponded to diverse, infrequent mutations, with fewer than 40 genes mutated in >2% of cases. In contrast, somatic structural variants, including new gene fusions and focal deletions of MBNL1, ZEB2 and ELF1, were disproportionately prevalent in young individuals as compared to adults. Conversely, mutations in DNMT3A and TP53, which were common in adults, were conspicuously absent from virtually all pediatric cases. New mutations in GATA2, FLT3 and CBL and recurrent mutations in MYC-ITD, NRAS, KRAS and WT1 were frequent in pediatric AML. Deletions, mutations and promoter DNA hypermethylation convergently impacted Wnt signaling, Polycomb repression, innate immune cell interactions and a cluster of zinc finger-encoding genes associated with KMT2A rearrangements. These results highlight the need for and facilitate the development of age-tailored targeted therapies for the treatment of pediatric AML.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Genomics in childhood acute myeloid leukemia comes of age.Nat Med. 2018 Jan 9;24(1):7-9. doi: 10.1038/nm.4469. Nat Med. 2018. PMID: 29315296 No abstract available.
Similar articles
-
[Characterization of mutational pattern of patients with core-binding factor acute myeloid leukemia].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2019 Jul 10;36(7):657-661. doi: 10.3760/cma.j.issn.1003-9406.2019.07.001. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2019. PMID: 31302905 Chinese.
-
Mutational landscape and clinical outcome of pediatric acute myeloid leukemia with 11q23/KMT2A rearrangements.Cancer Med. 2023 Jan;12(2):1418-1430. doi: 10.1002/cam4.5026. Epub 2022 Jul 14. Cancer Med. 2023. PMID: 35833755 Free PMC article.
-
What Is Abnormal in Normal Karyotype Acute Myeloid Leukemia in Children? Analysis of the Mutational Landscape and Prognosis of the TARGET-AML Cohort.Genes (Basel). 2021 May 21;12(6):792. doi: 10.3390/genes12060792. Genes (Basel). 2021. PMID: 34064268 Free PMC article.
-
[Genetic aberrations and new treatment strategies for pediatric acute myeloid leukemia].Rinsho Ketsueki. 2018;59(10):2260-2267. doi: 10.11406/rinketsu.59.2260. Rinsho Ketsueki. 2018. PMID: 30305534 Review. Japanese.
-
Comprehensive Mutation Profile in Acute Myeloid Leukemia Patients with RUNX1-RUNX1T1 or CBFB-MYH11 Fusions.Turk J Haematol. 2022 Jun 1;39(2):84-93. doi: 10.4274/tjh.galenos.2022.2021.0641. Epub 2022 Apr 21. Turk J Haematol. 2022. PMID: 35445594 Free PMC article. Review.
Cited by
-
NUP98::NSD1 and FLT3/ITD co-expression is an independent predictor of poor prognosis in pediatric AML patients.BMC Pediatr. 2024 Aug 24;24(1):547. doi: 10.1186/s12887-024-05007-3. BMC Pediatr. 2024. PMID: 39182032 Free PMC article.
-
UBTF Tandem Duplications in Pediatric MDS and AML: Implications for Clinical Screening and Diagnosis.medRxiv [Preprint]. 2023 Nov 13:2023.11.13.23298320. doi: 10.1101/2023.11.13.23298320. medRxiv. 2023. Update in: Haematologica. 2024 Aug 01;109(8):2459-2468. doi: 10.3324/haematol.2023.284683 PMID: 38014207 Free PMC article. Updated. Preprint.
-
Analytical study of RUNX1-RUNXT1, PML-RARA, CBFB-MYH11, BCR-ABL1p210 , and KMT2-MLLT3 in Mexican children with acute myeloid leukemia: A multicenter study of the Mexican interinstitutional group for the identification of the causes of childhood leukemia (MIGICCL).Front Pediatr. 2022 Nov 14;10:946690. doi: 10.3389/fped.2022.946690. eCollection 2022. Front Pediatr. 2022. PMID: 36452349 Free PMC article.
-
Differential sensitivity of acute myeloid leukemia cells to daunorubicin depends on P2X7A versus P2X7B receptor expression.Cell Death Dis. 2020 Oct 18;11(10):876. doi: 10.1038/s41419-020-03058-9. Cell Death Dis. 2020. PMID: 33071281 Free PMC article.
-
How Genetics and Genomics Advances Are Rewriting Pediatric Cancer Research and Clinical Care.Medicina (Kaunas). 2022 Oct 2;58(10):1386. doi: 10.3390/medicina58101386. Medicina (Kaunas). 2022. PMID: 36295546 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
