

BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics
- PMID: 29220515
- PMCID: PMC5850278
- DOI: 10.1093/molbev/msx319
BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics
Abstract
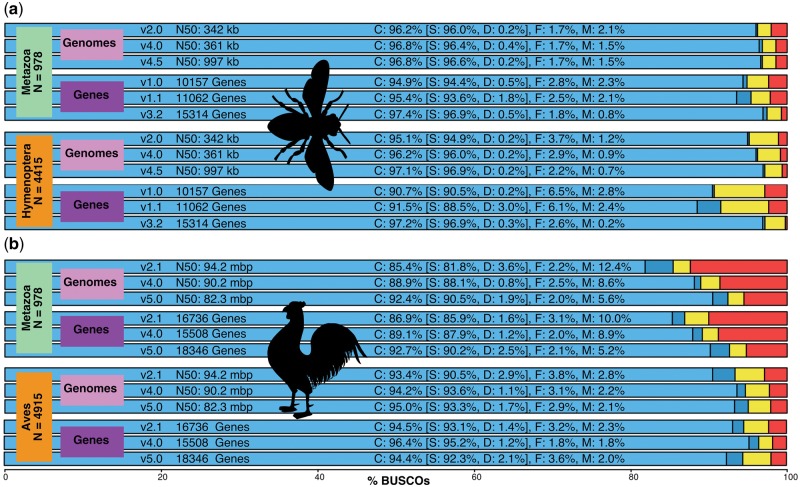
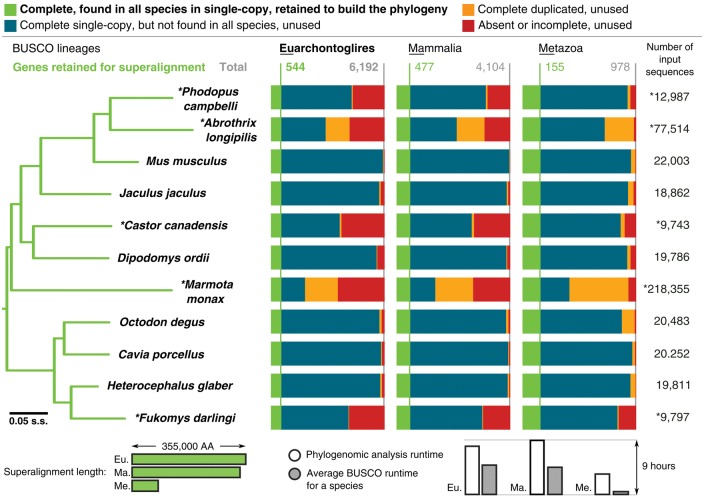
Genomics promises comprehensive surveying of genomes and metagenomes, but rapidly changing technologies and expanding data volumes make evaluation of completeness a challenging task. Technical sequencing quality metrics can be complemented by quantifying completeness of genomic data sets in terms of the expected gene content of Benchmarking Universal Single-Copy Orthologs (BUSCO, http://busco.ezlab.org). The latest software release implements a complete refactoring of the code to make it more flexible and extendable to facilitate high-throughput assessments. The original six lineage assessment data sets have been updated with improved species sampling, 34 new subsets have been built for vertebrates, arthropods, fungi, and prokaryotes that greatly enhance resolution, and data sets are now also available for nematodes, protists, and plants. Here, we present BUSCO v3 with example analyses that highlight the wide-ranging utility of BUSCO assessments, which extend beyond quality control of genomics data sets to applications in comparative genomics analyses, gene predictor training, metagenomics, and phylogenomics.
Keywords: bioinformatics; evolution; metagenomics; transcriptomics.
The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures

Similar articles
-
BUSCO: Assessing Genomic Data Quality and Beyond.Curr Protoc. 2021 Dec;1(12):e323. doi: 10.1002/cpz1.323. Curr Protoc. 2021. PMID: 34936221
-
BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs.Bioinformatics. 2015 Oct 1;31(19):3210-2. doi: 10.1093/bioinformatics/btv351. Epub 2015 Jun 9. Bioinformatics. 2015. PMID: 26059717
-
BUSCO: Assessing Genome Assembly and Annotation Completeness.Methods Mol Biol. 2019;1962:227-245. doi: 10.1007/978-1-4939-9173-0_14. Methods Mol Biol. 2019. PMID: 31020564
-
An Experimental Approach to Genome Annotation: This report is based on a colloquium sponsored by the American Academy of Microbiology held July 19-20, 2004, in Washington, DC.Washington (DC): American Society for Microbiology; 2004. Washington (DC): American Society for Microbiology; 2004. PMID: 33001599 Free Books & Documents. Review.
-
Next-generation sequencing technologies and their impact on microbial genomics.Brief Funct Genomics. 2013 Sep;12(5):440-53. doi: 10.1093/bfgp/els062. Epub 2013 Jan 11. Brief Funct Genomics. 2013. PMID: 23314033 Review.
Cited by
-
Semi-automated assembly of high-quality diploid human reference genomes.Nature. 2022 Nov;611(7936):519-531. doi: 10.1038/s41586-022-05325-5. Epub 2022 Oct 19. Nature. 2022. PMID: 36261518 Free PMC article.
-
New alignment-based sequence extraction software (ALiBaSeq) and its utility for deep level phylogenetics.PeerJ. 2021 Mar 31;9:e11019. doi: 10.7717/peerj.11019. eCollection 2021. PeerJ. 2021. PMID: 33850647 Free PMC article.
-
The gut ileal mucosal virome is disturbed in patients with Crohn's disease and exacerbates intestinal inflammation in mice.Nat Commun. 2024 Feb 22;15(1):1638. doi: 10.1038/s41467-024-45794-y. Nat Commun. 2024. PMID: 38388538 Free PMC article.
-
The role of introgression and ecotypic parallelism in delineating intraspecific conservation units.Mol Ecol. 2020 Aug;29(15):2793-2809. doi: 10.1111/mec.15522. Epub 2020 Jul 11. Mol Ecol. 2020. PMID: 32567754 Free PMC article.
-
Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird-of-paradise.Mol Ecol Resour. 2021 Jan;21(1):263-286. doi: 10.1111/1755-0998.13252. Epub 2020 Oct 10. Mol Ecol Resour. 2021. PMID: 32937018 Free PMC article.
References
-
- Davey JW, Chouteau M, Barker SL, Maroja L, Baxter SW, Simpson F, Joron M, Mallet J, Dasmahapatra KK, Jiggins CD.. 2016. Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 million years of butterfly evolution. G3 (Bethesda) 6(3):695–708. - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
