

Cardiac Insulin Signaling Regulates Glycolysis Through Phosphofructokinase 2 Content and Activity
- PMID: 29203581
- PMCID: PMC5779029
- DOI: 10.1161/JAHA.117.007159
Cardiac Insulin Signaling Regulates Glycolysis Through Phosphofructokinase 2 Content and Activity
Abstract
Background: The healthy heart has a dynamic capacity to respond and adapt to changes in nutrient availability. Diabetes mellitus disrupts this metabolic flexibility and promotes cardiomyopathy through mechanisms that are not completely understood. Phosphofructokinase 2 (PFK-2) is a primary regulator of cardiac glycolysis and substrate selection, yet its regulation under normal and pathological conditions is unknown. This study was undertaken to determine how changes in insulin signaling affect PFK-2 content, activity, and cardiac metabolism.
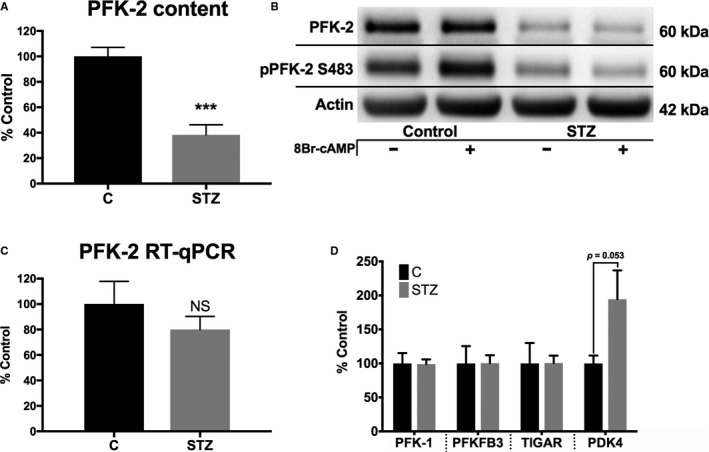
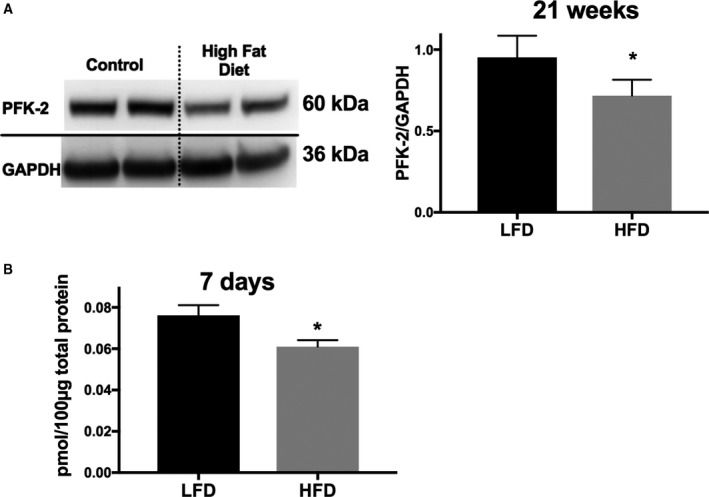
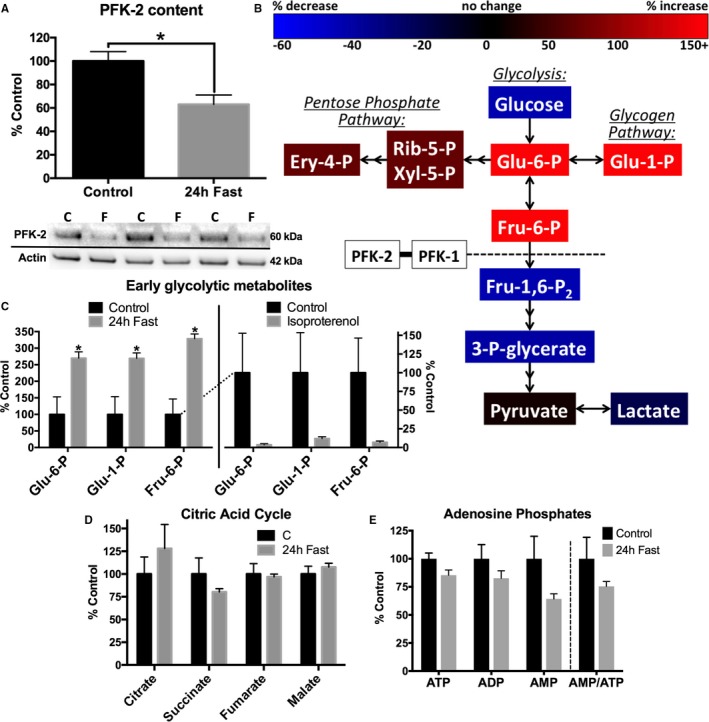
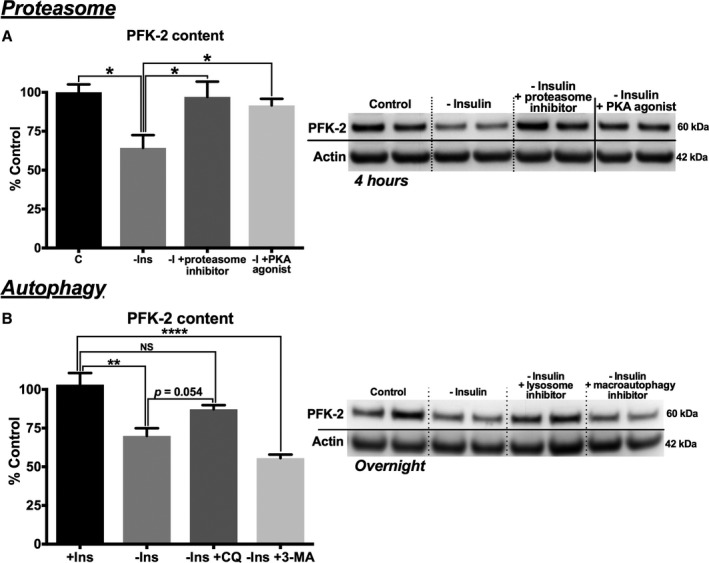
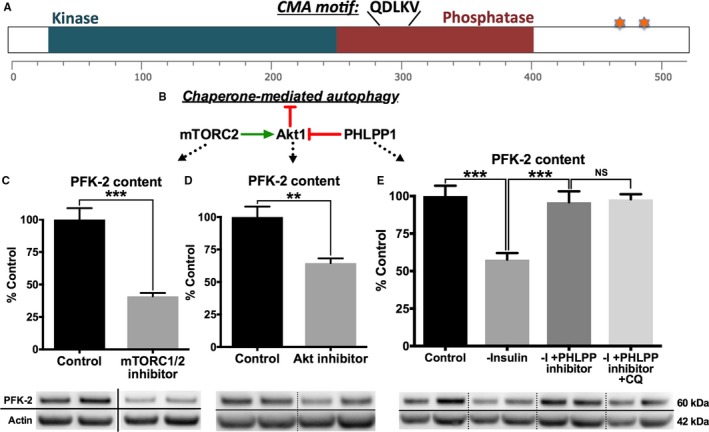
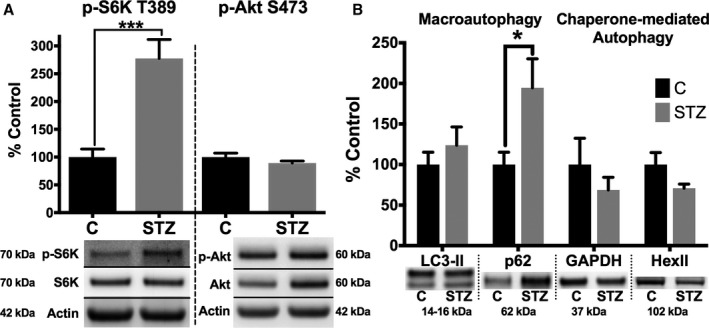
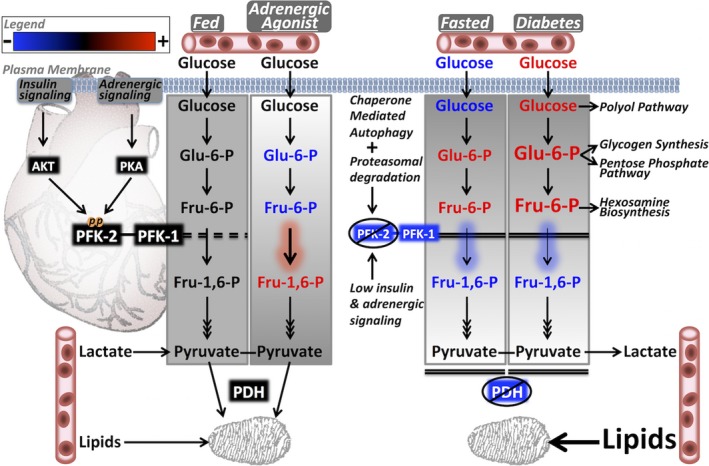
Methods and results: Streptozotocin-induced diabetes mellitus, high-fat diet feeding, and fasted mice were used to identify how decreased insulin signaling affects PFK-2 and cardiac metabolism. Primary adult cardiomyocytes were used to define the mechanisms that regulate PFK-2 degradation. Both type 1 diabetes mellitus and a high-fat diet induced a significant decrease in cardiac PFK-2 protein content without affecting its transcript levels. Overnight fasting also induced a decrease in PFK-2, suggesting it is rapidly degraded in the absence of insulin signaling. An unbiased metabolomic study demonstrated that decreased PFK-2 in fasted animals is accompanied by an increase in glycolytic intermediates upstream of phosphofructokianse-1, whereas those downstream are diminished. Mechanistic studies using cardiomyocytes showed that, in the absence of insulin signaling, PFK-2 is rapidly degraded via both proteasomal- and chaperone-mediated autophagy.
Conclusions: The loss of PFK-2 content as a result of reduced insulin signaling impairs the capacity to dynamically regulate glycolysis and elevates the levels of early glycolytic intermediates. Although this may be beneficial in the fasted state to conserve systemic glucose, it represents a pathological impairment in diabetes mellitus.
Keywords: autophagy; diabetic cardiomyopathy; glycolysis; insulin action; insulin resistance; metabolism.
© 2017 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley.
Figures

Similar articles
-
Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity.J Biol Chem. 2004 Nov 12;279(46):48085-90. doi: 10.1074/jbc.M405510200. Epub 2004 Aug 25. J Biol Chem. 2004. PMID: 15331593
-
Cardiac metabolism in a new rat model of type 2 diabetes using high-fat diet with low dose streptozotocin.Cardiovasc Diabetol. 2013 Sep 24;12:136. doi: 10.1186/1475-2840-12-136. Cardiovasc Diabetol. 2013. PMID: 24063408 Free PMC article.
-
Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice.Clin Sci (Lond). 2016 Apr;130(8):625-41. doi: 10.1042/CS20150623. Epub 2016 Jan 21. Clin Sci (Lond). 2016. PMID: 26795437
-
Insulin and ischemia stimulate glycolysis by acting on the same targets through different and opposing signaling pathways.J Mol Cell Cardiol. 2002 Sep;34(9):1091-7. doi: 10.1006/jmcc.2002.2063. J Mol Cell Cardiol. 2002. PMID: 12392881 Review.
-
Studies of gene expression and activity of hexokinase, phosphofructokinase and glycogen synthase in human skeletal muscle in states of altered insulin-stimulated glucose metabolism.Dan Med Bull. 1999 Feb;46(1):13-34. Dan Med Bull. 1999. PMID: 10081651 Review.
Cited by
-
Enhancing cardiac glycolysis causes an increase in PDK4 content in response to short-term high-fat diet.J Biol Chem. 2019 Nov 8;294(45):16831-16845. doi: 10.1074/jbc.RA119.010371. Epub 2019 Sep 27. J Biol Chem. 2019. PMID: 31562244 Free PMC article.
-
Sod1 integrates oxygen availability to redox regulate NADPH production and the thiol redoxome.Proc Natl Acad Sci U S A. 2022 Jan 4;119(1):e2023328119. doi: 10.1073/pnas.2023328119. Proc Natl Acad Sci U S A. 2022. PMID: 34969852 Free PMC article.
-
The role of glycolytic metabolic pathways in cardiovascular disease and potential therapeutic approaches.Basic Res Cardiol. 2023 Nov 8;118(1):48. doi: 10.1007/s00395-023-01018-w. Basic Res Cardiol. 2023. PMID: 37938421 Free PMC article. Review.
-
Research Progress on the Interaction Between Autophagy and Energy Homeostasis in Cardiac Remodeling.Front Pharmacol. 2020 Nov 30;11:587438. doi: 10.3389/fphar.2020.587438. eCollection 2020. Front Pharmacol. 2020. PMID: 33328993 Free PMC article. Review.
-
The role of glucose metabolism and insulin resistance in cardiac remodelling induced by cigarette smoke exposure.J Cell Mol Med. 2021 Jan;25(2):1314-1318. doi: 10.1111/jcmm.16053. Epub 2020 Dec 9. J Cell Mol Med. 2021. PMID: 33300293 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
