

Immunohaemostasis: a new view on haemostasis during sepsis
- PMID: 29197958
- PMCID: PMC5712298
- DOI: 10.1186/s13613-017-0339-5
Immunohaemostasis: a new view on haemostasis during sepsis
Abstract
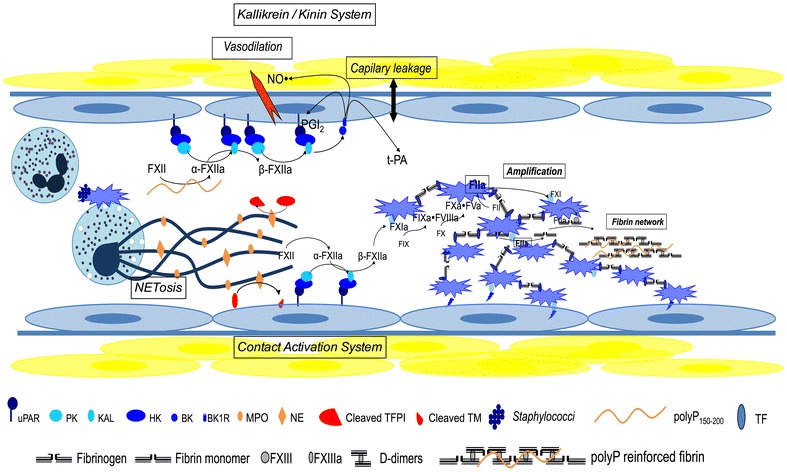
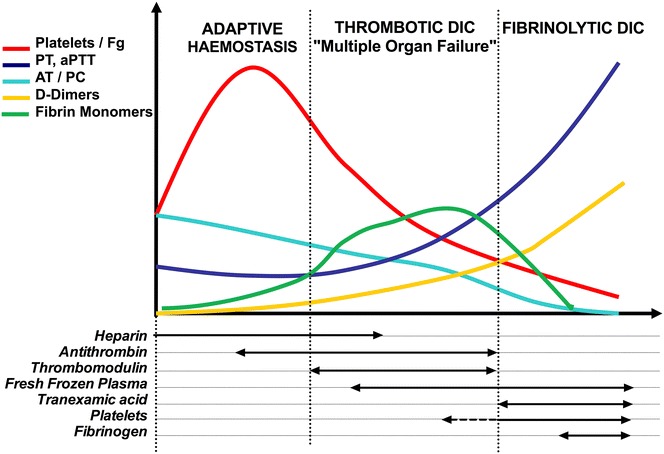
Host infection by a micro-organism triggers systemic inflammation, innate immunity and complement pathways, but also haemostasis activation. The role of thrombin and fibrin generation in host defence is now recognised, and thrombin has become a partner for survival, while it was seen only as one of the "principal suspects" of multiple organ failure and death during septic shock. This review is first focused on pathophysiology. The role of contact activation system, polyphosphates and neutrophil extracellular traps has emerged, offering new potential therapeutic targets. Interestingly, newly recognised host defence peptides (HDPs), derived from thrombin and other "coagulation" factors, are potent inhibitors of bacterial growth. Inhibition of thrombin generation could promote bacterial growth, while HDPs could become novel therapeutic agents against pathogens when resistance to conventional therapies grows. In a second part, we focused on sepsis-induced coagulopathy diagnostic challenge and stratification from "adaptive" haemostasis to "noxious" disseminated intravascular coagulation (DIC) either thrombotic or haemorrhagic. Besides usual coagulation tests, we discussed cellular haemostasis assessment including neutrophil, platelet and endothelial cell activation. Then, we examined therapeutic opportunities to prevent or to reduce "excess" thrombin generation, while preserving "adaptive" haemostasis. The fail of international randomised trials involving anticoagulants during septic shock may modify the hypothesis considering the end of haemostasis as a target to improve survival. On the one hand, patients at low risk of mortality may not be treated to preserve "immunothrombosis" as a defence when, on the other hand, patients at high risk with patent excess thrombin and fibrin generation could benefit from available (antithrombin, soluble thrombomodulin) or ongoing (FXI and FXII inhibitors) therapies. We propose to better assess coagulation response during infection by an improved knowledge of pathophysiology and systematic testing including determination of DIC scores. This is one of the clues to allocate the right treatment for the right patient at the right moment.
Keywords: Contact phase; Disseminated intravascular coagulation (DIC); Host defence peptides (HDPs); Infection; Neutrophil extracellular traps (NETs); Septic shock.
Figures
Similar articles
-
Normal prothrombinase activity, increased systemic thrombin activity, and lower antithrombin levels in patients with disseminated intravascular coagulation at an early phase of trauma: comparison with acute coagulopathy of trauma-shock.Surgery. 2013 Jul;154(1):48-57. doi: 10.1016/j.surg.2013.02.004. Epub 2013 May 16. Surgery. 2013. PMID: 23684364
-
The pathophysiology, diagnosis, and management of sepsis-associated disseminated intravascular coagulation.J Intensive Care. 2023 May 23;11(1):24. doi: 10.1186/s40560-023-00672-5. J Intensive Care. 2023. PMID: 37221630 Free PMC article. Review.
-
International Society on Thrombosis and Haemostasis score for overt disseminated intravascular coagulation predicts organ dysfunction and fatality in sepsis patients.Blood Coagul Fibrinolysis. 2006 Sep;17(6):445-51. doi: 10.1097/01.mbc.0000240916.63521.2e. Blood Coagul Fibrinolysis. 2006. PMID: 16905947
-
Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation: What We Need to Know and How to Manage for Prolonged Casualty Care.J Spec Oper Med. 2023 Jun 23;23(2):118-121. doi: 10.55460/6OZC-JIOV. J Spec Oper Med. 2023. PMID: 37302145 Review.
-
Trauma-induced innate immune activation and disseminated intravascular coagulation.J Thromb Haemost. 2024 Feb;22(2):337-351. doi: 10.1016/j.jtha.2023.09.028. Epub 2023 Oct 8. J Thromb Haemost. 2024. PMID: 37816463 Review.
Cited by
-
Neutrophil extracellular traps mediate neuro-immunothrombosis.Neural Regen Res. 2024 Aug 1;19(8):1734-1740. doi: 10.4103/1673-5374.389625. Epub 2023 Dec 11. Neural Regen Res. 2024. PMID: 38103239 Free PMC article.
-
Understanding, assessing and treating immune, endothelial and haemostasis dysfunctions in bacterial sepsis.Intensive Care Med. 2024 Oct;50(10):1580-1592. doi: 10.1007/s00134-024-07586-2. Epub 2024 Sep 2. Intensive Care Med. 2024. PMID: 39222142 Review.
-
Decreasing Plasma Fibrinogen Levels in the Intensive Care Unit Are Associated with High Mortality Rates In Patients With Sepsis-Induced Coagulopathy.Clin Appl Thromb Hemost. 2022 Jan-Dec;28:10760296221101386. doi: 10.1177/10760296221101386. Clin Appl Thromb Hemost. 2022. PMID: 35549920 Free PMC article.
-
Sepsis-induced coagulopathy: a matter of timeline.Intensive Care Med. 2024 Aug;50(8):1404-1405. doi: 10.1007/s00134-024-07507-3. Epub 2024 Jun 10. Intensive Care Med. 2024. PMID: 38856751 No abstract available.
-
Current Understanding of How Extracorporeal Membrane Oxygenators Activate Haemostasis and Other Blood Components.Front Med (Lausanne). 2018 Dec 12;5:352. doi: 10.3389/fmed.2018.00352. eCollection 2018. Front Med (Lausanne). 2018. PMID: 30619862 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
