

Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data
- PMID: 29130882
- PMCID: PMC5718706
- DOI: 10.7554/eLife.26476
Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data
Abstract
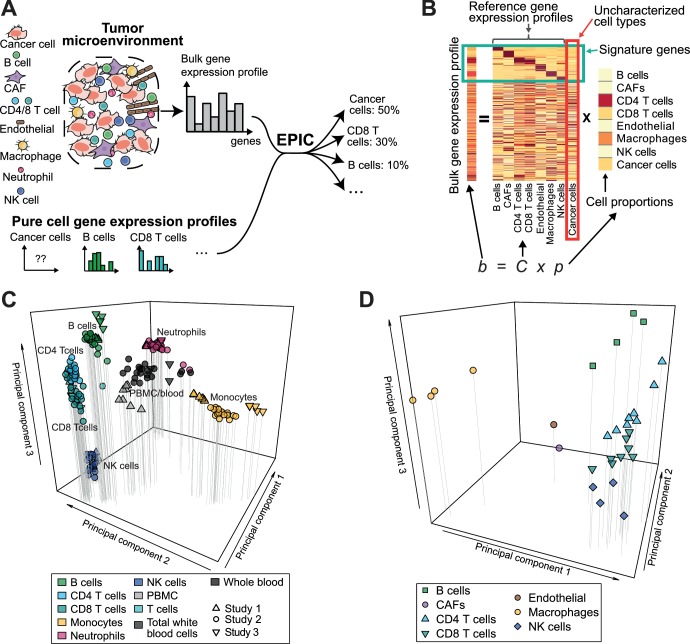
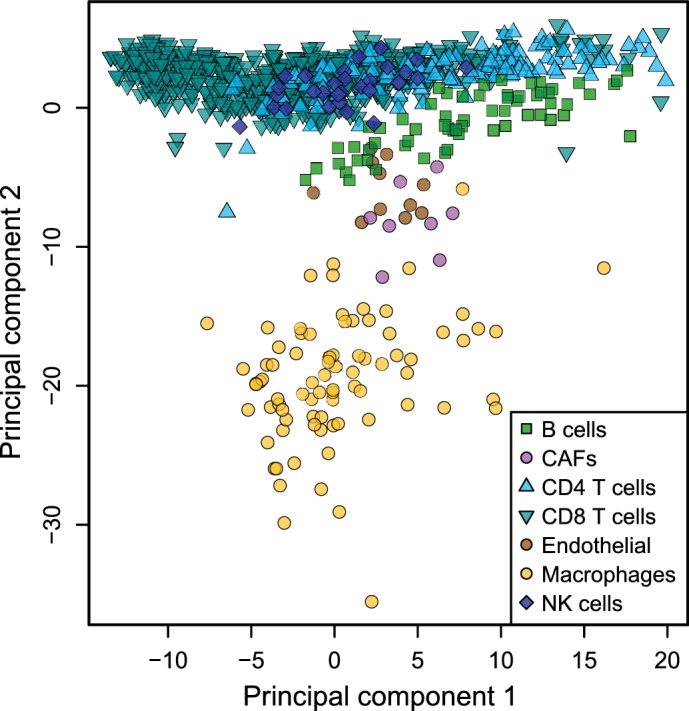
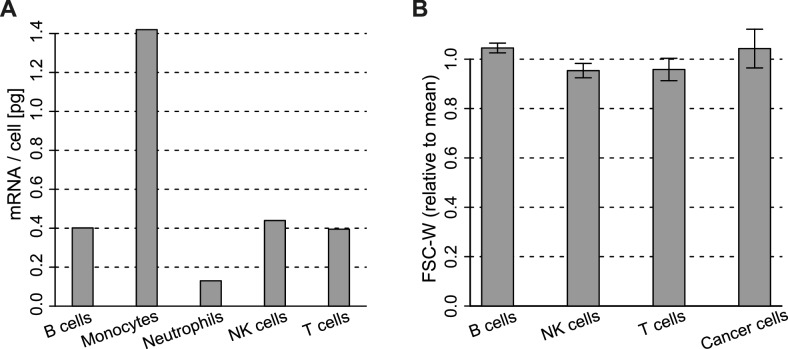
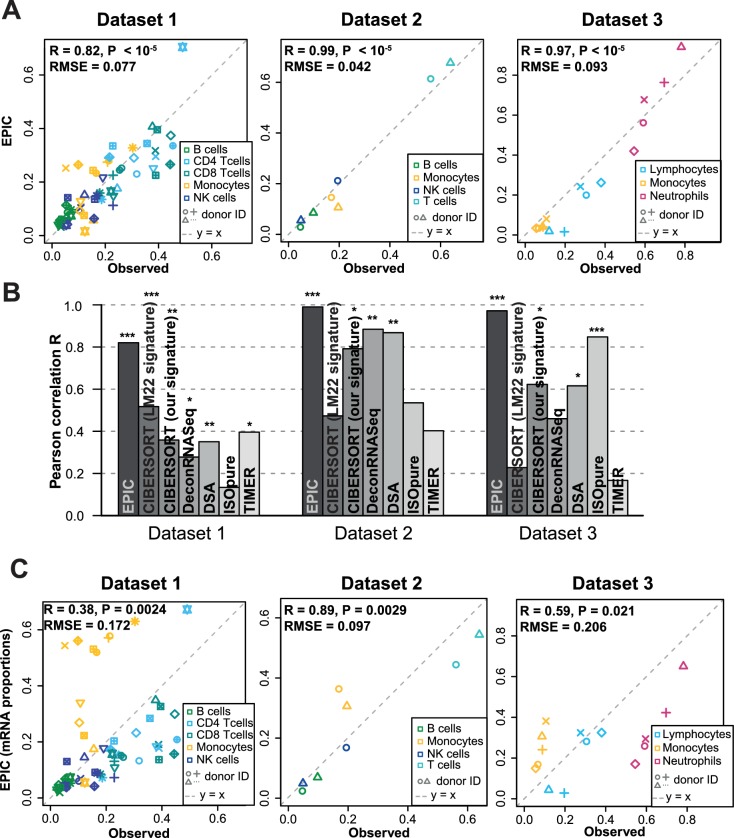
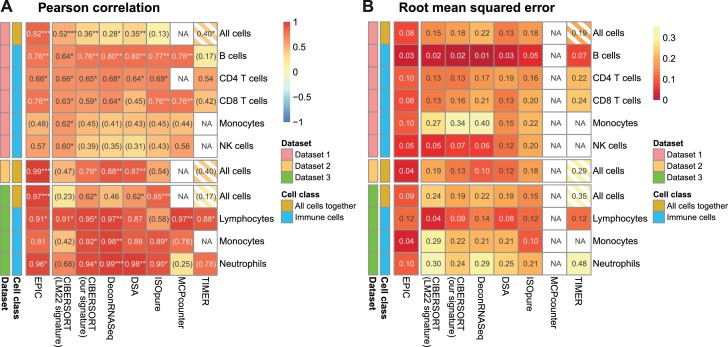
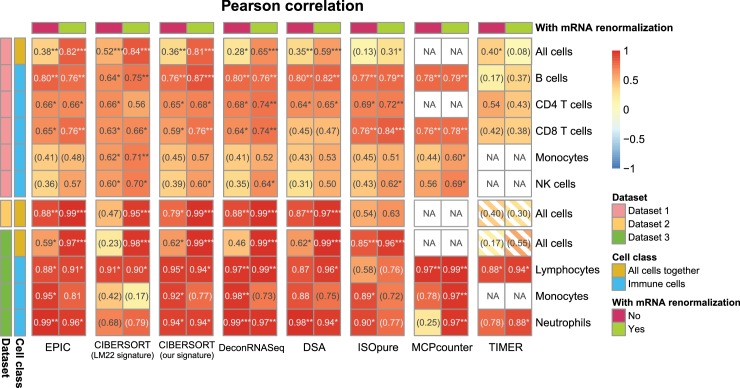
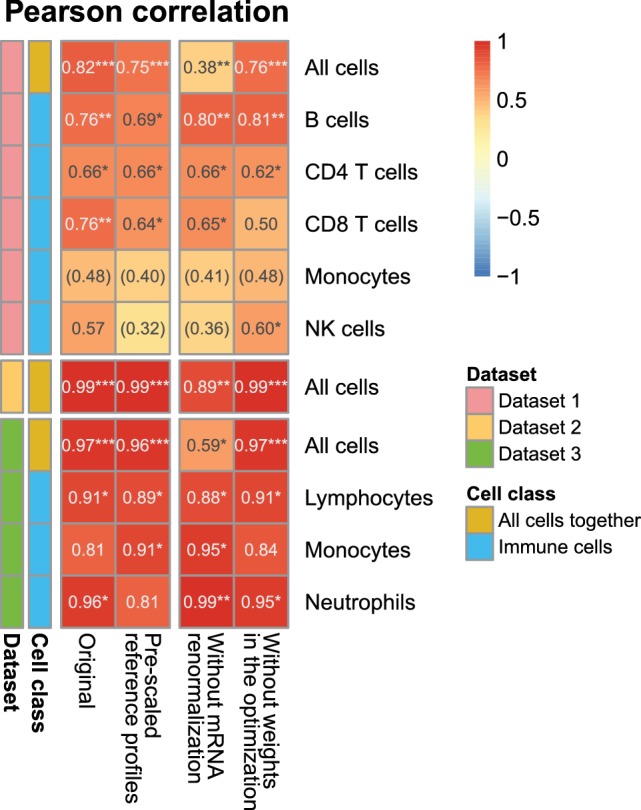
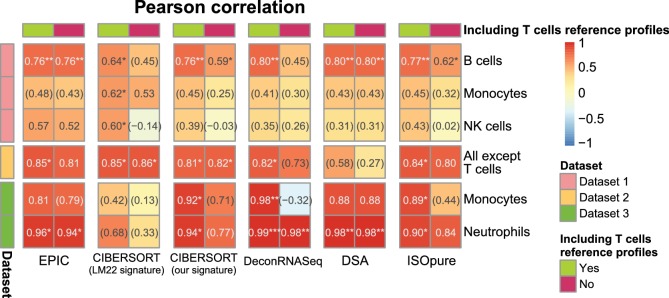
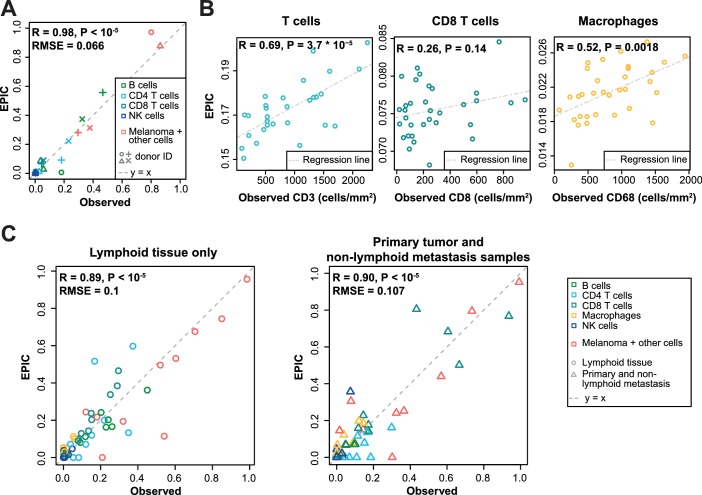
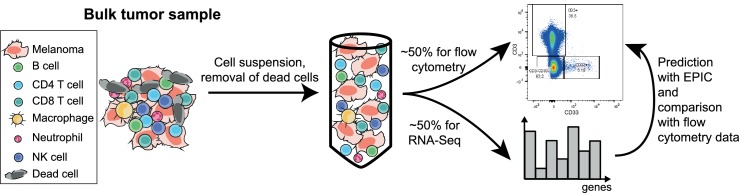
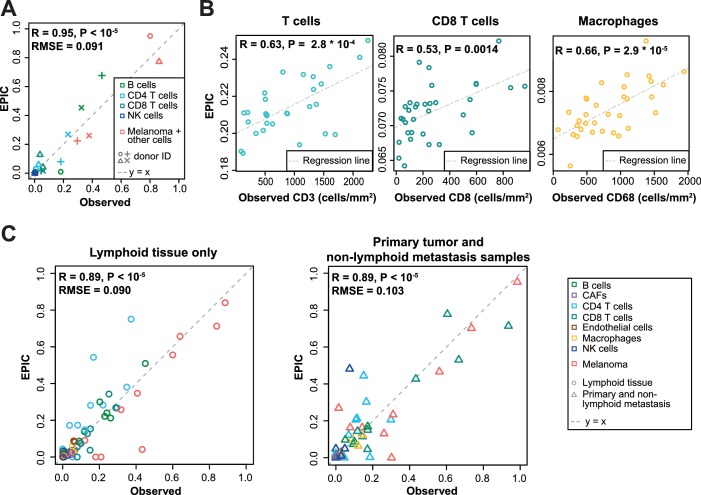
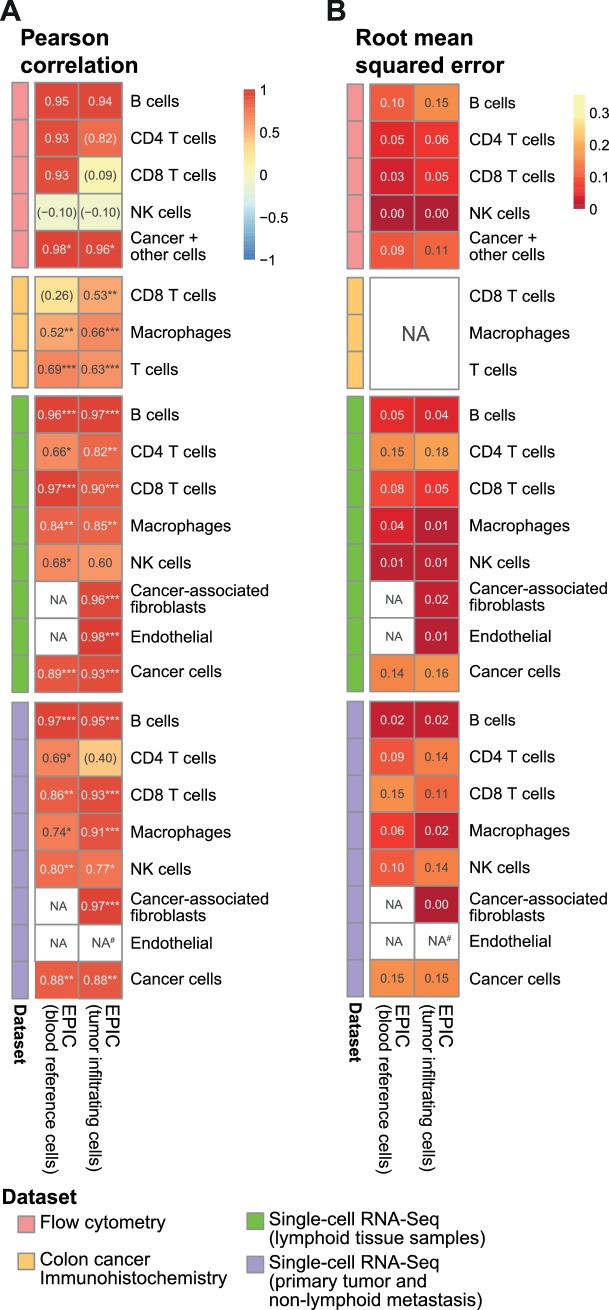
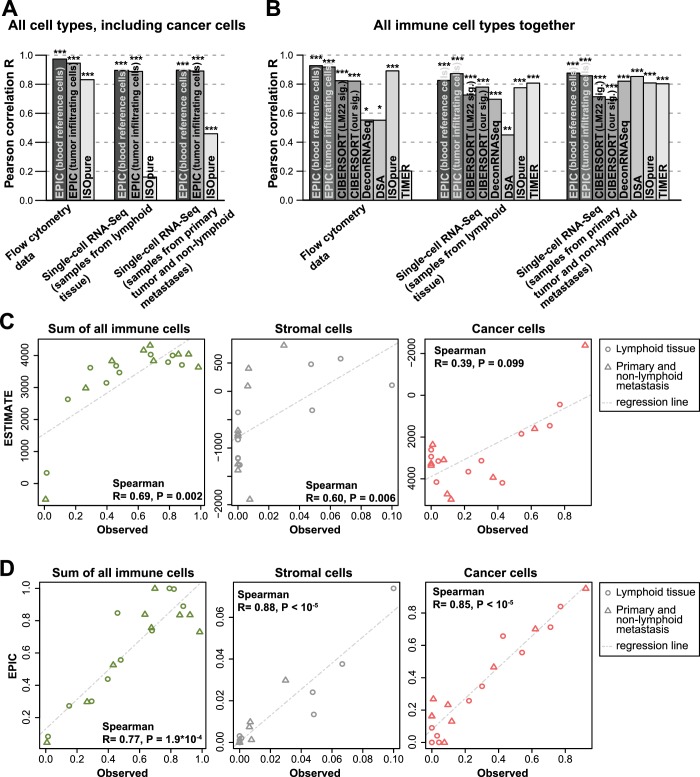
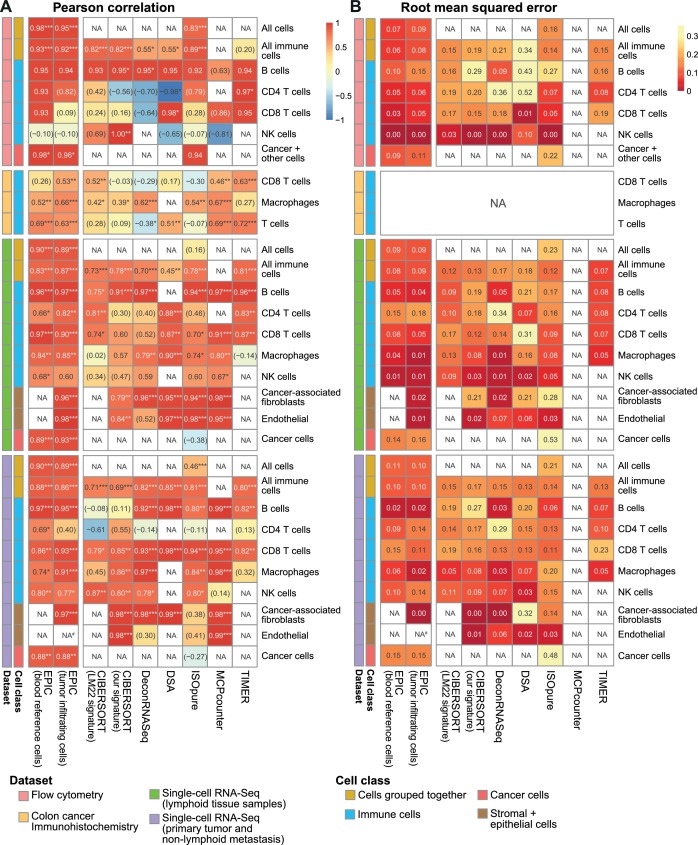
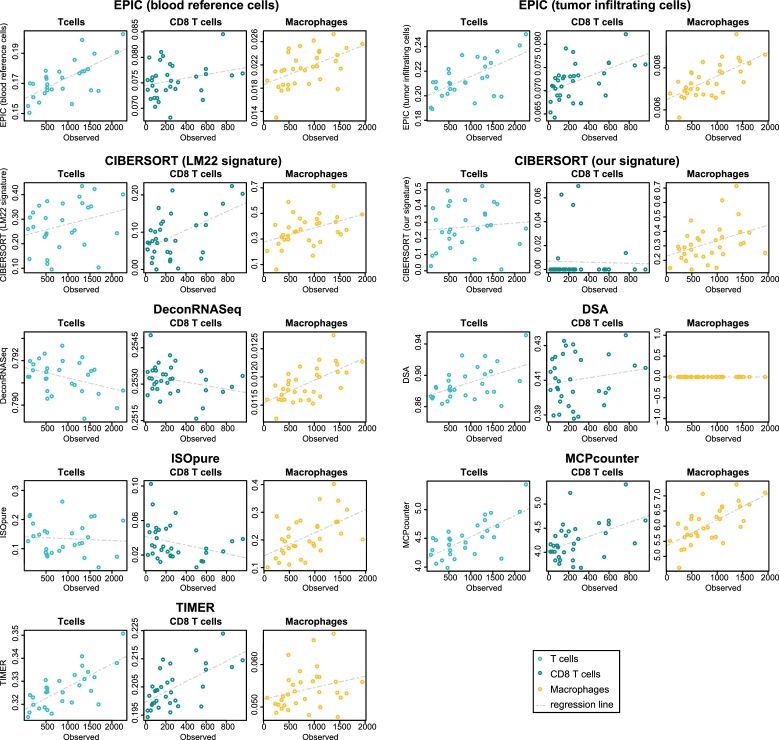
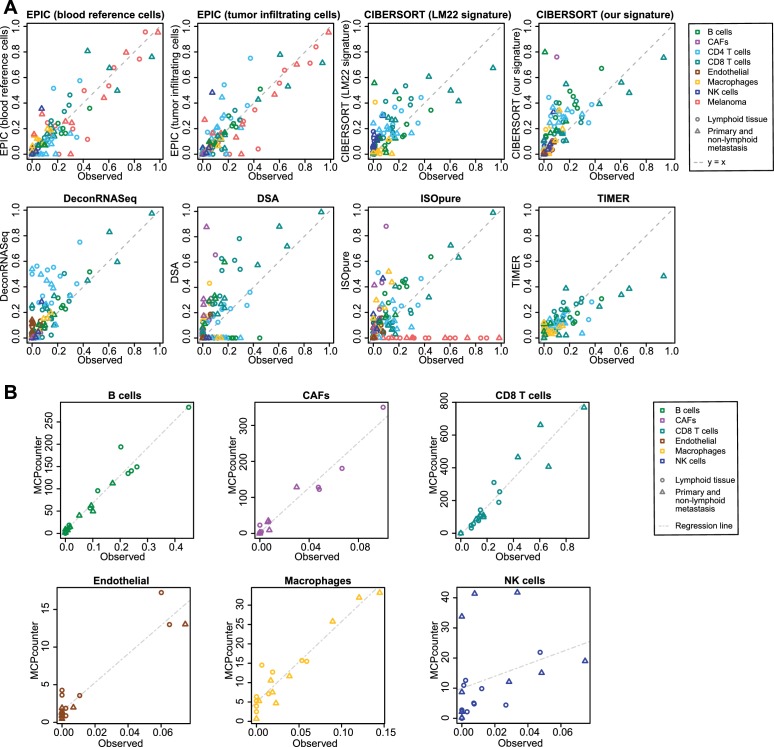
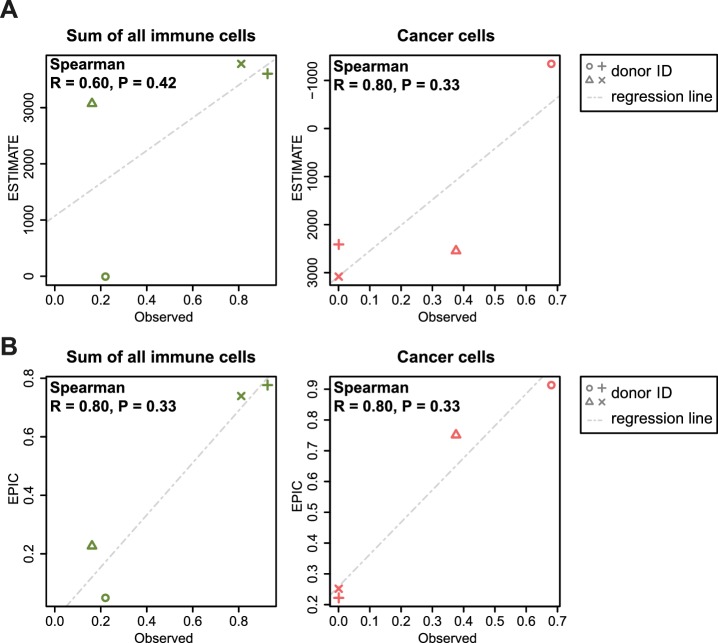
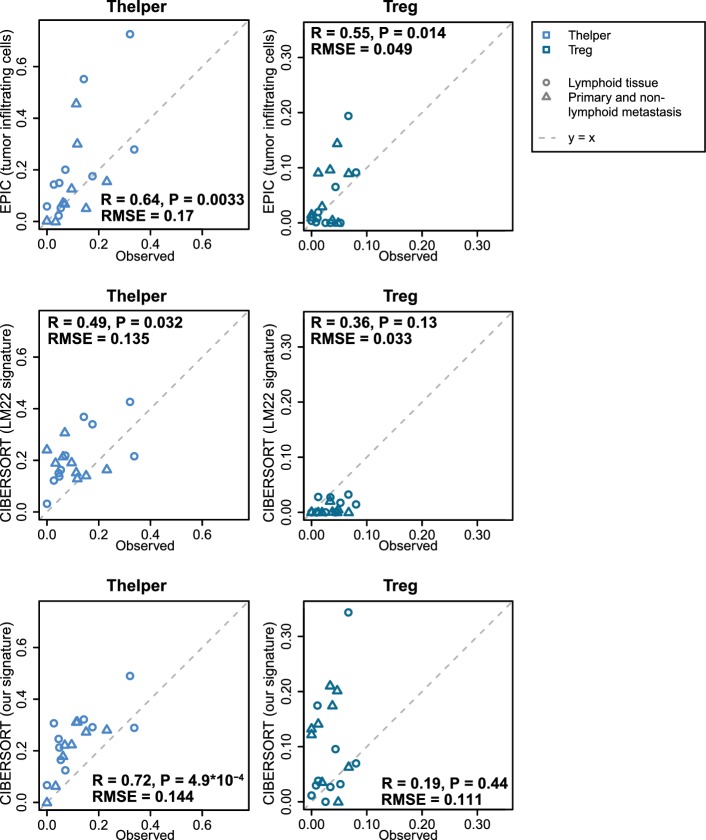
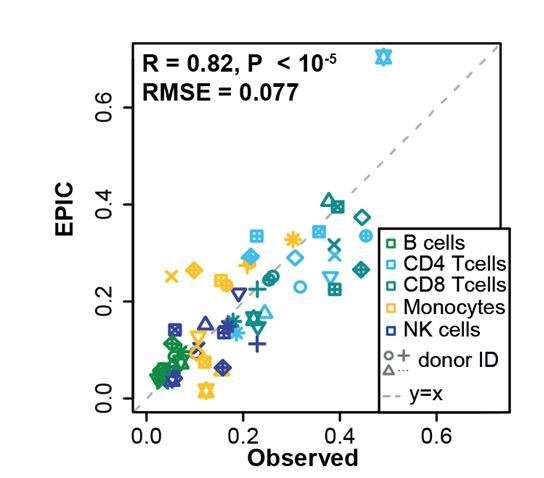
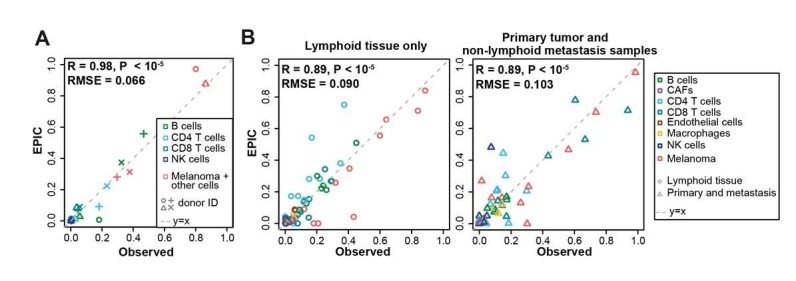
Immune cells infiltrating tumors can have important impact on tumor progression and response to therapy. We present an efficient algorithm to simultaneously estimate the fraction of cancer and immune cell types from bulk tumor gene expression data. Our method integrates novel gene expression profiles from each major non-malignant cell type found in tumors, renormalization based on cell-type-specific mRNA content, and the ability to consider uncharacterized and possibly highly variable cell types. Feasibility is demonstrated by validation with flow cytometry, immunohistochemistry and single-cell RNA-Seq analyses of human melanoma and colorectal tumor specimens. Altogether, our work not only improves accuracy but also broadens the scope of absolute cell fraction predictions from tumor gene expression data, and provides a unique novel experimental benchmark for immunogenomics analyses in cancer research (http://epic.gfellerlab.org).
Keywords: cancer biology; cell fraction predictions; computational biology; gene expression; human; systems biology; tumor immune microenvironment.
Conflict of interest statement
No competing interests declared.
Figures

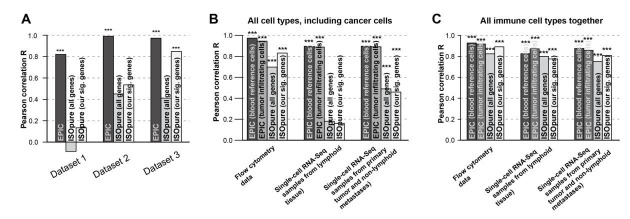
Similar articles
-
EPIC: A Tool to Estimate the Proportions of Different Cell Types from Bulk Gene Expression Data.Methods Mol Biol. 2020;2120:233-248. doi: 10.1007/978-1-0716-0327-7_17. Methods Mol Biol. 2020. PMID: 32124324
-
A Cancer-Specific Qualitative Method for Estimating the Proportion of Tumor-Infiltrating Immune Cells.Front Immunol. 2021 May 14;12:672031. doi: 10.3389/fimmu.2021.672031. eCollection 2021. Front Immunol. 2021. PMID: 34054849 Free PMC article.
-
Calculation of immune cell proportion from batch tumor gene expression profile based on support vector regression.J Bioinform Comput Biol. 2020 Oct;18(5):2050030. doi: 10.1142/S0219720020500304. Epub 2020 Aug 21. J Bioinform Comput Biol. 2020. PMID: 32825808
-
[Hopes and pitfalls of the molecular classification of breast cancer].Cesk Patol. 2015;51(1):26-32. Cesk Patol. 2015. PMID: 25671359 Review. Czech.
-
High-throughput genomic profiling of tumor-infiltrating leukocytes.Curr Opin Immunol. 2016 Aug;41:77-84. doi: 10.1016/j.coi.2016.06.006. Epub 2016 Jun 30. Curr Opin Immunol. 2016. PMID: 27372732 Free PMC article. Review.
Cited by
-
New HCC Subtypes Based on CD8 Tex-Related lncRNA Signature Could Predict Prognosis, Immunological and Drug Sensitivity Characteristics of Hepatocellular Carcinoma.J Hepatocell Carcinoma. 2024 Jul 5;11:1331-1355. doi: 10.2147/JHC.S459150. eCollection 2024. J Hepatocell Carcinoma. 2024. PMID: 38983937 Free PMC article.
-
Comparative analysis of somatic variant calling on matched FF and FFPE WGS samples.BMC Med Genomics. 2020 Jul 6;13(1):94. doi: 10.1186/s12920-020-00746-5. BMC Med Genomics. 2020. PMID: 32631411 Free PMC article.
-
Profiling of Lymphovascular Space Invasion in Cervical Cancer Revealed PI3K/Akt Signaling Pathway Overactivation and Heterogenic Tumor-Immune Microenvironments.Life (Basel). 2023 Dec 14;13(12):2342. doi: 10.3390/life13122342. Life (Basel). 2023. PMID: 38137942 Free PMC article.
-
Development and validation of prognostic models for colon adenocarcinoma based on combined immune-and metabolism-related genes.Front Oncol. 2022 Oct 31;12:1025397. doi: 10.3389/fonc.2022.1025397. eCollection 2022. Front Oncol. 2022. PMID: 36387195 Free PMC article.
-
Potential regulation and prognostic model of colorectal cancer with extracellular matrix genes.Heliyon. 2024 Aug 13;10(16):e36164. doi: 10.1016/j.heliyon.2024.e36164. eCollection 2024 Aug 30. Heliyon. 2024. PMID: 39247375 Free PMC article.
References
-
- Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, Trajanoski Z. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biology. 2015;16:64. doi: 10.1186/s13059-015-0620-6. - DOI - PMC - PubMed
-
- Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, Melton DA, Yanai A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Systems. 2016;3:346–360. doi: 10.1016/j.cels.2016.08.011. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
