

Foamy Virus Vector Carries a Strong Insulator in Its Long Terminal Repeat Which Reduces Its Genotoxic Potential
- PMID: 29046446
- PMCID: PMC5730768
- DOI: 10.1128/JVI.01639-17
Foamy Virus Vector Carries a Strong Insulator in Its Long Terminal Repeat Which Reduces Its Genotoxic Potential
Abstract
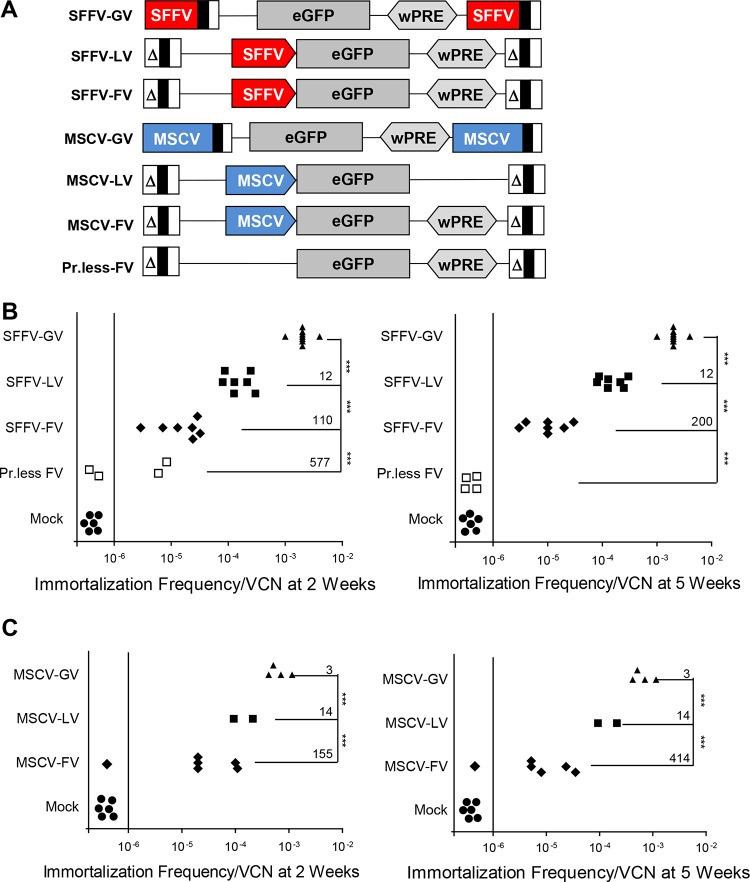
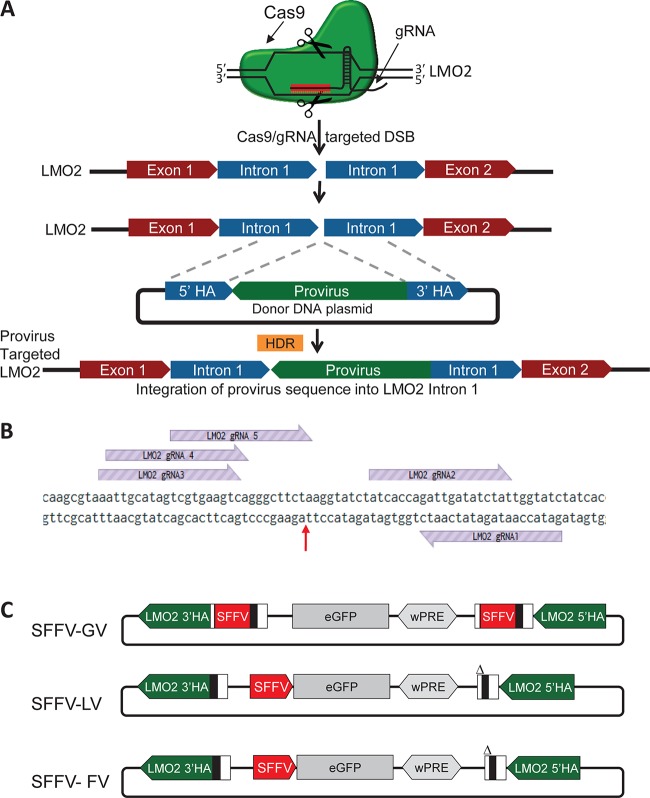
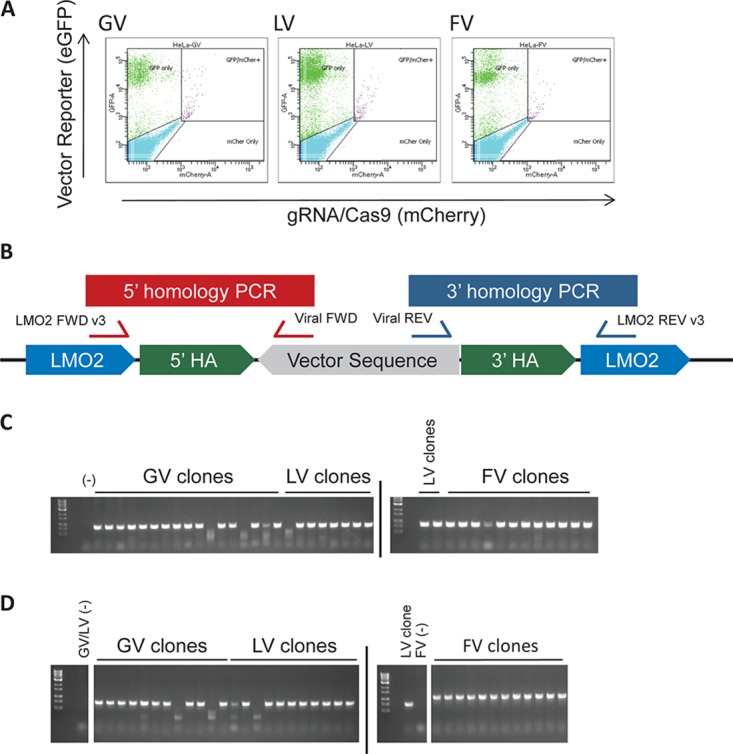
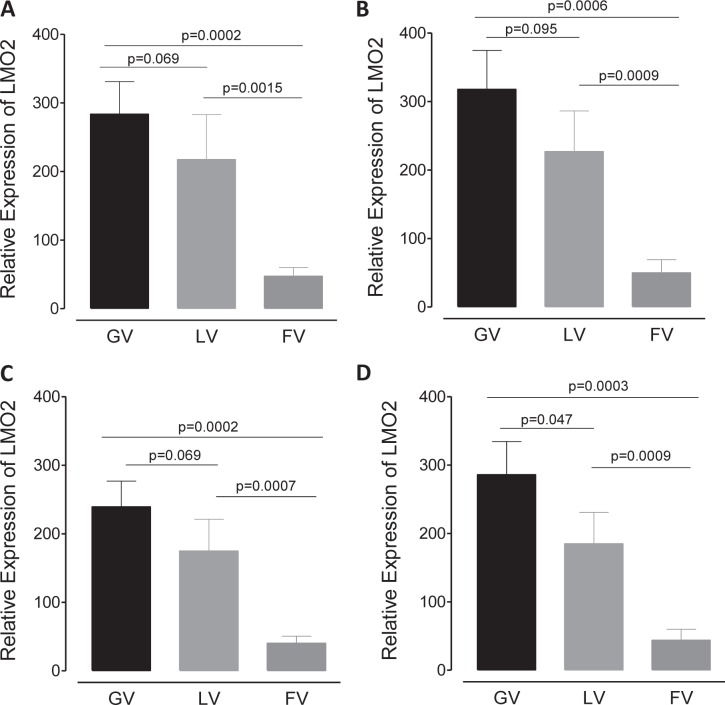
Strong viral enhancers in gammaretrovirus vectors have caused cellular proto-oncogene activation and leukemia, necessitating the use of cellular promoters in "enhancerless" self-inactivating integrating vectors. However, cellular promoters result in relatively low transgene expression, often leading to inadequate disease phenotype correction. Vectors derived from foamy virus, a nonpathogenic retrovirus, show higher preference for nongenic integrations than gammaretroviruses/lentiviruses and preferential integration near transcriptional start sites, like gammaretroviruses. We found that strong viral enhancers/promoters placed in foamy viral vectors caused extremely low immortalization of primary mouse hematopoietic stem/progenitor cells compared to analogous gammaretrovirus/lentivirus vectors carrying the same enhancers/promoters, an effect not explained solely by foamy virus' modest insertional site preference for nongenic regions compared to gammaretrovirus/lentivirus vectors. Using CRISPR/Cas9-mediated targeted insertion of analogous proviral sequences into the LMO2 gene and then measuring LMO2 expression, we demonstrate a sequence-specific effect of foamy virus, independent of insertional bias, contributing to reduced genotoxicity. We show that this effect is mediated by a 36-bp insulator located in the foamy virus long terminal repeat (LTR) that has high-affinity binding to the CCCTC-binding factor. Using our LMO2 activation assay, LMO2 expression was significantly increased when this insulator was removed from foamy virus and significantly reduced when the insulator was inserted into the lentiviral LTR. Our results elucidate a mechanism underlying the low genotoxicity of foamy virus, identify a novel insulator, and support the use of foamy virus as a vector for gene therapy, especially when strong enhancers/promoters are required.IMPORTANCE Understanding the genotoxic potential of viral vectors is important in designing safe and efficacious vectors for gene therapy. Self-inactivating vectors devoid of viral long-terminal-repeat enhancers have proven safe; however, transgene expression from cellular promoters is often insufficient for full phenotypic correction. Foamy virus is an attractive vector for gene therapy. We found foamy virus vectors to be remarkably less genotoxic, well below what was expected from their integration site preferences. We demonstrate that the foamy virus long terminal repeats contain an insulator element that binds CCCTC-binding factor and reduces its insertional genotoxicity. Our study elucidates a mechanism behind the low genotoxic potential of foamy virus, identifies a unique insulator, and supports the use of foamy virus as a vector for gene therapy.
Keywords: CCCTC-binding factor (CTCF); CRISPR/Cas; foamy virus; gene insulator; gene therapy; genotoxicity.
Copyright © 2017 American Society for Microbiology.
Figures

Similar articles
-
Insulated Foamy Viral Vectors.Hum Gene Ther. 2016 Mar;27(3):255-66. doi: 10.1089/hum.2015.110. Epub 2016 Mar 15. Hum Gene Ther. 2016. PMID: 26715244 Free PMC article.
-
Alpharetroviral self-inactivating vectors: long-term transgene expression in murine hematopoietic cells and low genotoxicity.Mol Ther. 2012 May;20(5):1022-32. doi: 10.1038/mt.2011.309. Epub 2012 Feb 14. Mol Ther. 2012. PMID: 22334016 Free PMC article.
-
Evaluating the Safety of Retroviral Vectors Based on Insertional Oncogene Activation and Blocked Differentiation in Cultured Thymocytes.Mol Ther. 2016 Jun;24(6):1090-1099. doi: 10.1038/mt.2016.55. Epub 2016 Mar 9. Mol Ther. 2016. PMID: 26957223 Free PMC article.
-
Viral Vectors: The Road to Reducing Genotoxicity.Toxicol Sci. 2017 Feb;155(2):315-325. doi: 10.1093/toxsci/kfw220. Epub 2016 Nov 1. Toxicol Sci. 2017. PMID: 27803388 Review.
-
Foamy virus vectors: an awaited alternative to gammaretro- and lentiviral vectors.Curr Gene Ther. 2007 Aug;7(4):261-71. doi: 10.2174/156652307781369092. Curr Gene Ther. 2007. PMID: 17969559 Review.
Cited by
-
Oncolytic Foamy Virus - generation and properties of a nonpathogenic replicating retroviral vector system that targets chronically proliferating cancer cells.J Virol. 2021 Apr 26;95(10):e00015-21. doi: 10.1128/JVI.00015-21. Epub 2021 Mar 10. J Virol. 2021. PMID: 33692205 Free PMC article.
-
Restoration of Functional Full-Length Dystrophin After Intramuscular Transplantation of Foamy Virus-Transduced Myoblasts.Hum Gene Ther. 2020 Feb;31(3-4):241-252. doi: 10.1089/hum.2019.224. Epub 2020 Jan 10. Hum Gene Ther. 2020. PMID: 31801386 Free PMC article.
-
FV Vectors as Alternative Gene Vehicles for Gene Transfer in HSCs.Viruses. 2020 Mar 19;12(3):332. doi: 10.3390/v12030332. Viruses. 2020. PMID: 32204324 Free PMC article. Review.
-
Molecular Epidemiology and Whole-Genome Analysis of Bovine Foamy Virus in Japan.Viruses. 2021 May 28;13(6):1017. doi: 10.3390/v13061017. Viruses. 2021. PMID: 34071542 Free PMC article.
-
The human leukemia virus HTLV-1 alters the structure and transcription of host chromatin in cis.Elife. 2018 Jun 26;7:e36245. doi: 10.7554/eLife.36245. Elife. 2018. PMID: 29941091 Free PMC article.
References
-
- Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, Martinache C, Rieux-Laucat F, Latour S, Belohradsky BH, Leiva L, Sorensen R, Debre M, Casanova JL, Blanche S, Durandy A, Bushman FD, Fischer A, Cavazzana-Calvo M. 2010. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 363:355–364. doi:10.1056/NEJMoa1000164. - DOI - PMC - PubMed
-
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. 2008. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 118:3132–3142. doi:10.1172/JCI35700. - DOI - PMC - PubMed
-
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJ, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB, Thrasher AJ. 2008. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest 118:3143–3150. doi:10.1172/JCI35798. - DOI - PMC - PubMed
-
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Gohring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kuhlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. 2010. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 16:198–204. doi:10.1038/nm.2088. - DOI - PubMed
-
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, Glimm H, Kuhlcke K, Schilz A, Kunkel H, Naundorf S, Brinkmann A, Deichmann A, Fischer M, Ball C, Pilz I, Dunbar C, Du Y, Jenkins NA, Copeland NG, Luthi U, Hassan M, Thrasher AJ, Hoelzer D, von Kalle C, Seger R, Grez M. 2006. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med 12:401–409. doi:10.1038/nm1393. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
