

Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis
- PMID: 29018272
- PMCID: PMC5899973
- DOI: 10.1038/nrgastro.2017.119
Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis
Abstract
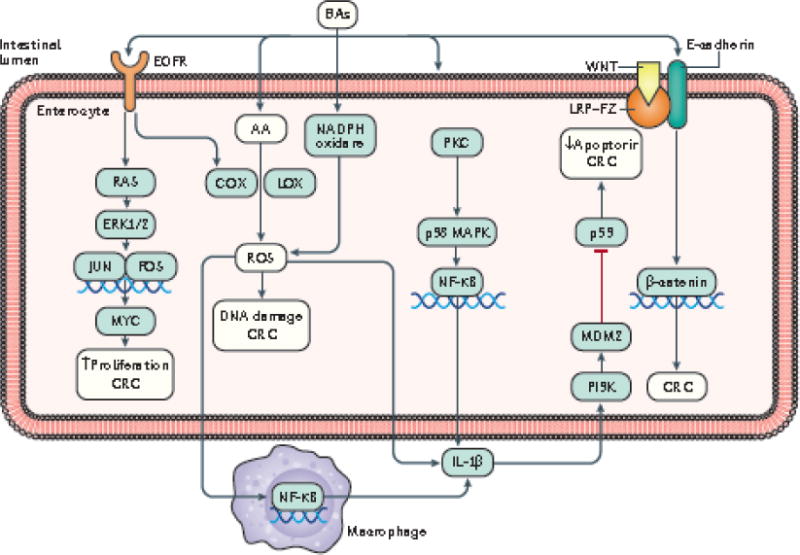
Emerging evidence points to a strong association between the gut microbiota and the risk, development and progression of gastrointestinal cancers such as colorectal cancer (CRC) and hepatocellular carcinoma (HCC). Bile acids, produced in the liver, are metabolized by enzymes derived from intestinal bacteria and are critically important for maintaining a healthy gut microbiota, balanced lipid and carbohydrate metabolism, insulin sensitivity and innate immunity. Given the complexity of bile acid signalling and the direct biochemical interactions between the gut microbiota and the host, a systems biology perspective is required to understand the liver-bile acid-microbiota axis and its role in gastrointestinal carcinogenesis to reverse the microbiota-mediated alterations in bile acid metabolism that occur in disease states. An examination of recent research progress in this area is urgently needed. In this Review, we discuss the mechanistic links between bile acids and gastrointestinal carcinogenesis in CRC and HCC, which involve two major bile acid-sensing receptors, farnesoid X receptor (FXR) and G protein-coupled bile acid receptor 1 (TGR5). We also highlight the strategies and cutting-edge technologies to target gut-microbiota-dependent alterations in bile acid metabolism in the context of cancer therapy.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Probiotics, bile acids and gastrointestinal carcinogenesis.Nat Rev Gastroenterol Hepatol. 2018 Apr;15(4):205. doi: 10.1038/nrgastro.2018.24. Epub 2018 Mar 7. Nat Rev Gastroenterol Hepatol. 2018. PMID: 29512647 Free PMC article. No abstract available.
-
Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis: a role for bifidobacteria and lactobacilli?Nat Rev Gastroenterol Hepatol. 2018 Apr;15(4):205. doi: 10.1038/nrgastro.2018.23. Epub 2018 Mar 7. Nat Rev Gastroenterol Hepatol. 2018. PMID: 29512648 No abstract available.
Similar articles
-
The gut microbiome-bile acid axis in hepatocarcinogenesis.Biomed Pharmacother. 2021 Jan;133:111036. doi: 10.1016/j.biopha.2020.111036. Epub 2020 Nov 28. Biomed Pharmacother. 2021. PMID: 33378947 Review.
-
The gut microbiota at the intersection of bile acids and intestinal carcinogenesis: An old story, yet mesmerizing.Int J Cancer. 2020 Apr 1;146(7):1780-1790. doi: 10.1002/ijc.32563. Epub 2019 Jul 23. Int J Cancer. 2020. PMID: 31291465 Review.
-
Gut Microbiota-Bile Acid Crosstalk in Diarrhea-Irritable Bowel Syndrome.Biomed Res Int. 2020 Nov 12;2020:3828249. doi: 10.1155/2020/3828249. eCollection 2020. Biomed Res Int. 2020. PMID: 33274207 Free PMC article. Review.
-
Bile Acid Modifications at the Microbe-Host Interface: Potential for Nutraceutical and Pharmaceutical Interventions in Host Health.Annu Rev Food Sci Technol. 2016;7:313-33. doi: 10.1146/annurev-food-041715-033159. Epub 2016 Jan 11. Annu Rev Food Sci Technol. 2016. PMID: 26772409 Review.
-
Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism.Hepatology. 2018 Oct;68(4):1574-1588. doi: 10.1002/hep.29857. Epub 2018 May 21. Hepatology. 2018. PMID: 29486523 Free PMC article.
Cited by
-
Effects of bile acids on the growth, composition and metabolism of gut bacteria.NPJ Biofilms Microbiomes. 2024 Oct 23;10(1):112. doi: 10.1038/s41522-024-00566-w. NPJ Biofilms Microbiomes. 2024. PMID: 39438471 Free PMC article.
-
G protein-coupled receptors as potential targets for nonalcoholic fatty liver disease treatment.World J Gastroenterol. 2021 Feb 28;27(8):677-691. doi: 10.3748/wjg.v27.i8.677. World J Gastroenterol. 2021. PMID: 33716447 Free PMC article. Review.
-
Gut microbiota-mitochondrial inter-talk in non-alcoholic fatty liver disease.Front Nutr. 2022 Sep 20;9:934113. doi: 10.3389/fnut.2022.934113. eCollection 2022. Front Nutr. 2022. PMID: 36204383 Free PMC article. Review.
-
Antibiotic-Induced Changes in Microbiome-Related Metabolites and Bile Acids in Rat Plasma.Metabolites. 2020 Jun 11;10(6):242. doi: 10.3390/metabo10060242. Metabolites. 2020. PMID: 32545183 Free PMC article.
-
Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites.Gut. 2021 Apr;70(4):761-774. doi: 10.1136/gutjnl-2019-319664. Epub 2020 Jul 21. Gut. 2021. PMID: 32694178 Free PMC article.
References
-
- Mucci LA, Wedren S, Tamimi RM, Trichopoulos D, Adami HO. The role of gene-environment interaction in the aetiology of human cancer: examples from cancers of the large bowel, lung and breast. J Intern Med. 2001;249:477–493. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. - PubMed
-
- American Cancer Society. About Colorectal Cancer. 2017 https://www.cancer.org/content/dam/CRC/PDF/Public/8604.00.pdf.
-
- International Agency for Research on Cancer. GLOBOCAN 2012: Estimated Incidence, Mortality and Prevalence Worldwide in 2012. 2012 http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx.
-
- Jemal A, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
