

Biochemical and cellular properties of insulin receptor signalling
- PMID: 28974775
- PMCID: PMC5894887
- DOI: 10.1038/nrm.2017.89
Biochemical and cellular properties of insulin receptor signalling
Abstract
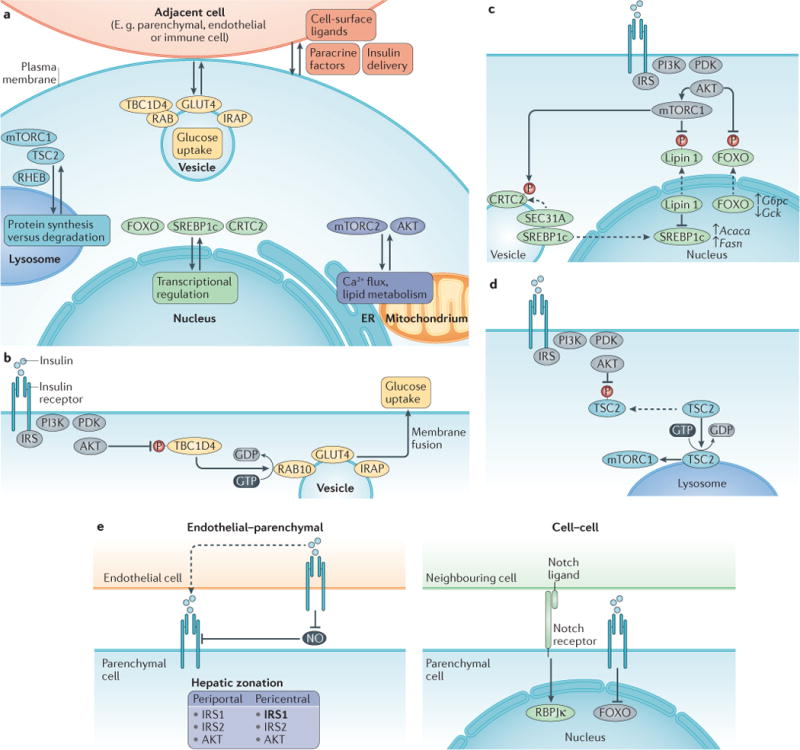
The mechanism of insulin action is a central theme in biology and medicine. In addition to the rather rare condition of insulin deficiency caused by autoimmune destruction of pancreatic β-cells, genetic and acquired abnormalities of insulin action underlie the far more common conditions of type 2 diabetes, obesity and insulin resistance. The latter predisposes to diseases ranging from hypertension to Alzheimer disease and cancer. Hence, understanding the biochemical and cellular properties of insulin receptor signalling is arguably a priority in biomedical research. In the past decade, major progress has led to the delineation of mechanisms of glucose transport, lipid synthesis, storage and mobilization. In addition to direct effects of insulin on signalling kinases and metabolic enzymes, the discovery of mechanisms of insulin-regulated gene transcription has led to a reassessment of the general principles of insulin action. These advances will accelerate the discovery of new treatment modalities for diabetes.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Metabolism and insulin signaling in common metabolic disorders and inherited insulin resistance.Dan Med J. 2014 Jul;61(7):B4890. Dan Med J. 2014. PMID: 25123125 Review.
-
Akt phosphorylates insulin receptor substrate to limit PI3K-mediated PIP3 synthesis.Elife. 2021 Jul 13;10:e66942. doi: 10.7554/eLife.66942. Elife. 2021. PMID: 34253290 Free PMC article.
-
Hyperinsulinaemia in cancer.Nat Rev Cancer. 2020 Nov;20(11):629-644. doi: 10.1038/s41568-020-0295-5. Epub 2020 Sep 9. Nat Rev Cancer. 2020. PMID: 32908223 Review.
-
Modulation of insulin action.Diabetologia. 2004 Feb;47(2):170-84. doi: 10.1007/s00125-003-1313-3. Epub 2004 Jan 13. Diabetologia. 2004. PMID: 14722654 Review.
-
Molecular mechanisms of insulin receptor substrate protein-mediated modulation of insulin signalling.FEBS Lett. 2003 Jul 3;546(1):32-6. doi: 10.1016/s0014-5793(03)00438-1. FEBS Lett. 2003. PMID: 12829233 Review.
Cited by
-
The impact of the gut microbiota on the reproductive and metabolic endocrine system.Gut Microbes. 2021 Jan-Dec;13(1):1-21. doi: 10.1080/19490976.2021.1894070. Gut Microbes. 2021. PMID: 33722164 Free PMC article. Review.
-
Activation of the insulin receptor by an insulin mimetic peptide.Nat Commun. 2022 Sep 23;13(1):5594. doi: 10.1038/s41467-022-33274-0. Nat Commun. 2022. PMID: 36151101 Free PMC article.
-
Orphan receptor GPR50 attenuates inflammation and insulin signaling in 3T3-L1 preadipocytes.FEBS Open Bio. 2023 Jan;13(1):89-101. doi: 10.1002/2211-5463.13516. Epub 2022 Dec 13. FEBS Open Bio. 2023. PMID: 36333974 Free PMC article.
-
MiT/TFE factors control ER-phagy via transcriptional regulation of FAM134B.EMBO J. 2020 Sep 1;39(17):e105696. doi: 10.15252/embj.2020105696. Epub 2020 Jul 27. EMBO J. 2020. PMID: 32716134 Free PMC article.
-
Andrographolide Promotes Uptake of Glucose and GLUT4 Transport through the PKC Pathway in L6 Cells.Pharmaceuticals (Basel). 2022 Oct 31;15(11):1346. doi: 10.3390/ph15111346. Pharmaceuticals (Basel). 2022. PMID: 36355518 Free PMC article.
References
-
- Levine R, Goldstein MS, Huddlestun B, Klein SP. Action of insulin on the ‘permeability’ of cells to free hexoses, as studied by its effect on the distribution of galactose. Am J Physiol. 1950;163:70–76. - PubMed
-
- Kasuga M, Zick Y, Blithe DL, Crettaz M, Kahn CR. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature. 1982;298:667–669. - PubMed
-
- Ebina Y, et al. The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling. Cell. 1985;40:747–758. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
