

Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours
- PMID: 28953875
- PMCID: PMC6050590
- DOI: 10.1038/nature24028
Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours
Erratum in
-
Author Correction: Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours.Nature. 2018 Jun;558(7710):E1. doi: 10.1038/s41586-018-0111-5. Nature. 2018. PMID: 29769713
Abstract
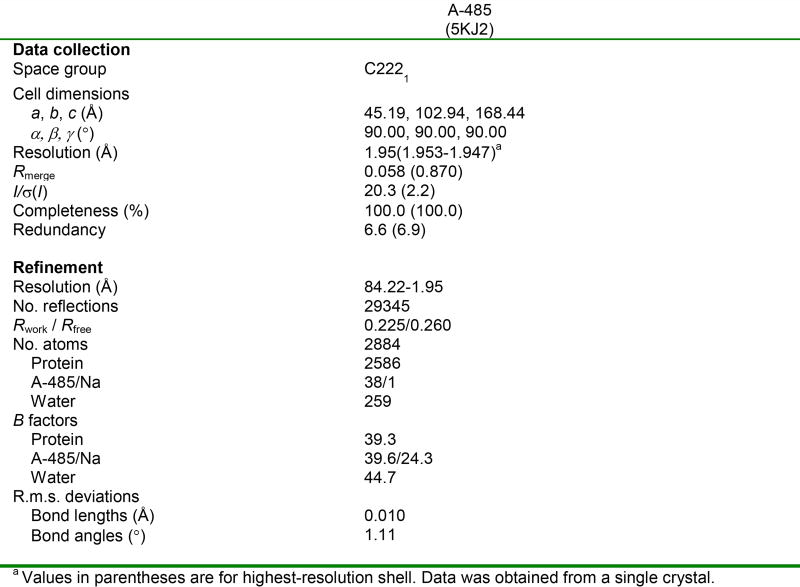
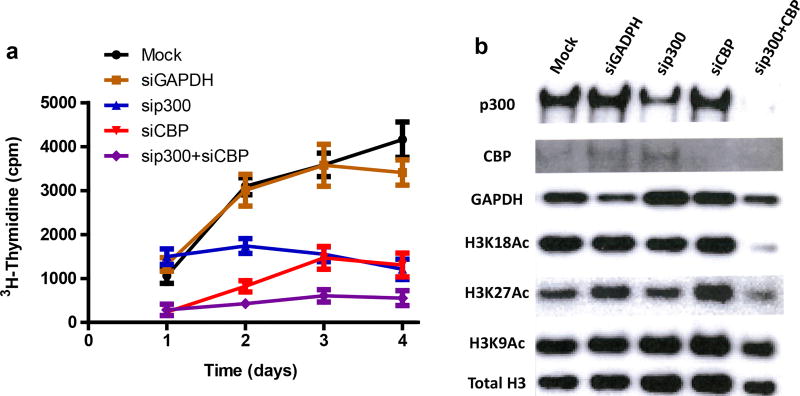
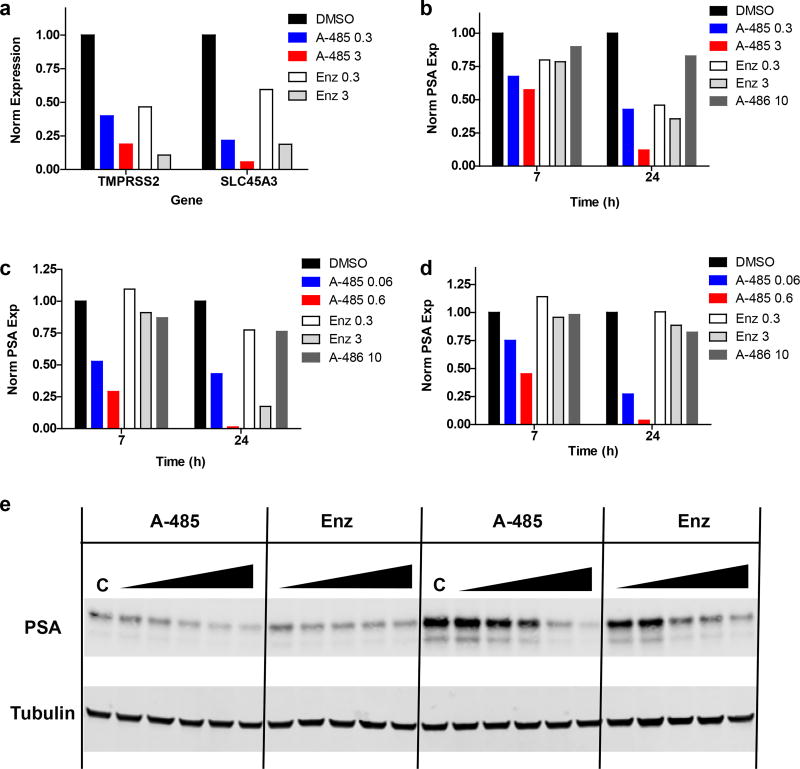
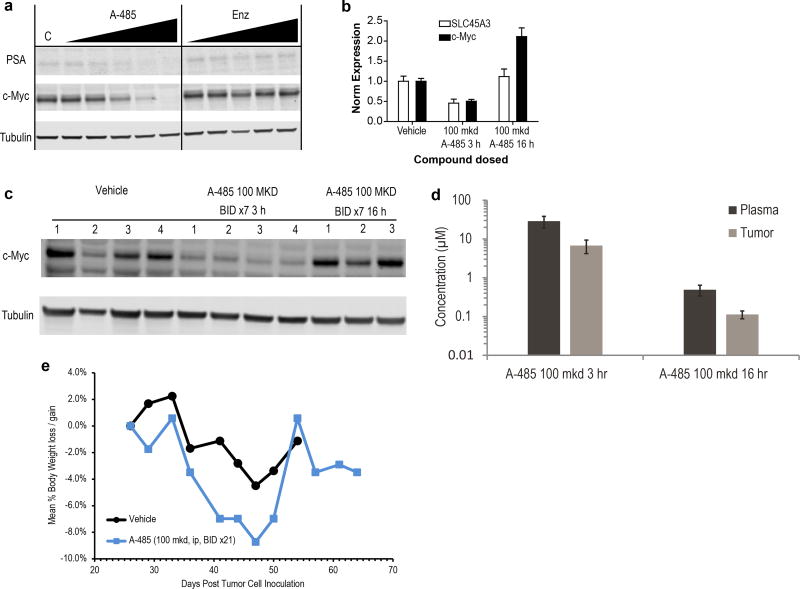
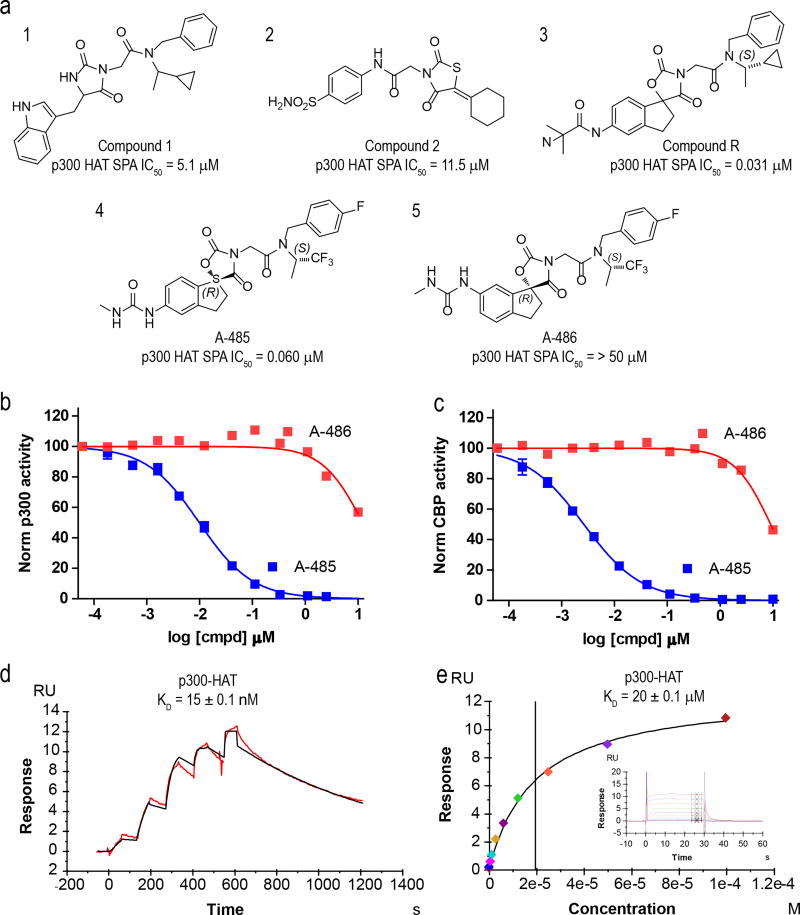
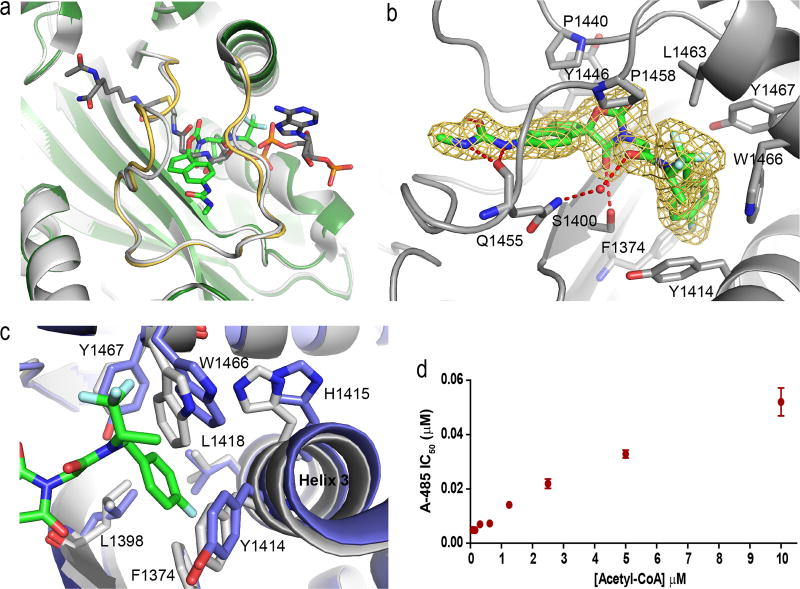
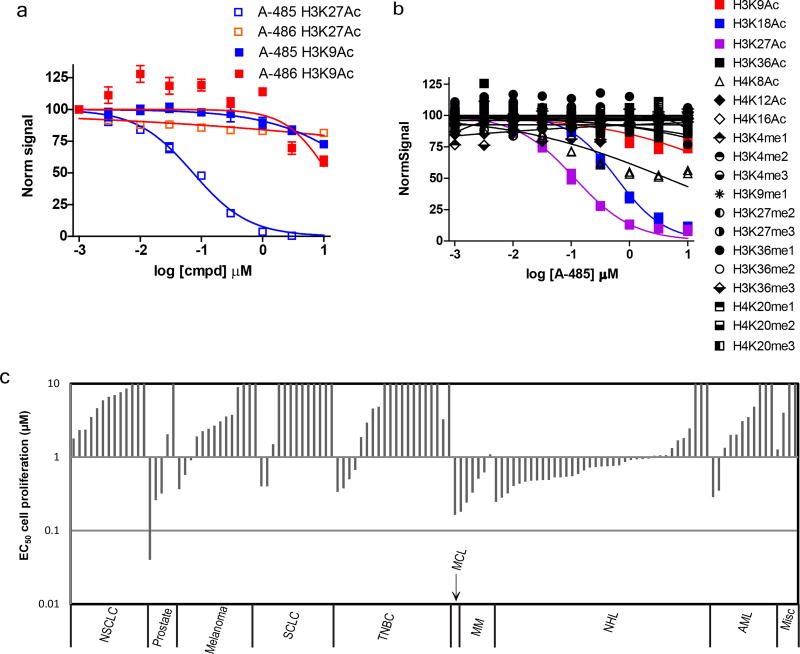
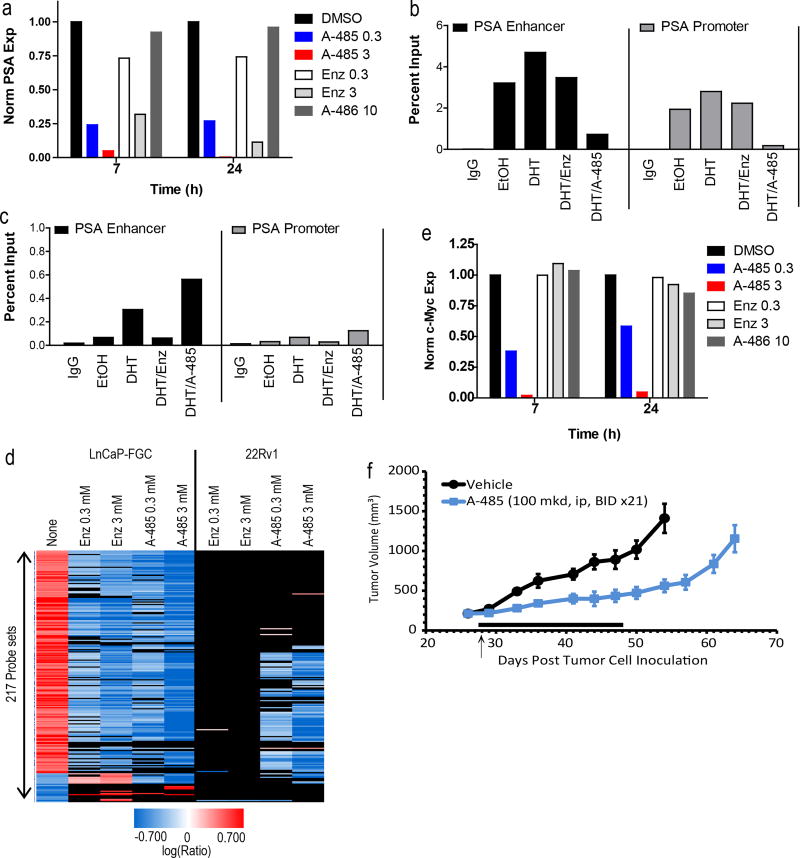
The dynamic and reversible acetylation of proteins, catalysed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), is a major epigenetic regulatory mechanism of gene transcription and is associated with multiple diseases. Histone deacetylase inhibitors are currently approved to treat certain cancers, but progress on the development of drug-like histone actyltransferase inhibitors has lagged behind. The histone acetyltransferase paralogues p300 and CREB-binding protein (CBP) are key transcriptional co-activators that are essential for a multitude of cellular processes, and have also been implicated in human pathological conditions (including cancer). Current inhibitors of the p300 and CBP histone acetyltransferase domains, including natural products, bi-substrate analogues and the widely used small molecule C646, lack potency or selectivity. Here, we describe A-485, a potent, selective and drug-like catalytic inhibitor of p300 and CBP. We present a high resolution (1.95 Å) co-crystal structure of a small molecule bound to the catalytic active site of p300 and demonstrate that A-485 competes with acetyl coenzyme A (acetyl-CoA). A-485 selectively inhibited proliferation in lineage-specific tumour types, including several haematological malignancies and androgen receptor-positive prostate cancer. A-485 inhibited the androgen receptor transcriptional program in both androgen-sensitive and castration-resistant prostate cancer and inhibited tumour growth in a castration-resistant xenograft model. These results demonstrate the feasibility of using small molecule inhibitors to selectively target the catalytic activity of histone acetyltransferases, which may provide effective treatments for transcriptional activator-driven malignancies and diseases.
Figures






Similar articles
-
Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP.Biochemistry. 2019 Apr 23;58(16):2133-2143. doi: 10.1021/acs.biochem.9b00160. Epub 2019 Apr 11. Biochemistry. 2019. PMID: 30924641 Free PMC article.
-
Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor.Chem Biol. 2010 May 28;17(5):471-82. doi: 10.1016/j.chembiol.2010.03.006. Chem Biol. 2010. PMID: 20534345 Free PMC article.
-
Current development of CBP/p300 inhibitors in the last decade.Eur J Med Chem. 2021 Jan 1;209:112861. doi: 10.1016/j.ejmech.2020.112861. Epub 2020 Oct 1. Eur J Med Chem. 2021. PMID: 33045661 Review.
-
The structural basis of protein acetylation by the p300/CBP transcriptional coactivator.Nature. 2008 Feb 14;451(7180):846-50. doi: 10.1038/nature06546. Nature. 2008. PMID: 18273021
-
Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function.Curr Opin Struct Biol. 2008 Dec;18(6):741-7. doi: 10.1016/j.sbi.2008.09.004. Epub 2008 Oct 27. Curr Opin Struct Biol. 2008. PMID: 18845255 Free PMC article. Review.
Cited by
-
CBP/p300 and HDAC activities regulate H3K27 acetylation dynamics and zygotic genome activation in mouse preimplantation embryos.EMBO J. 2022 Nov 17;41(22):e112012. doi: 10.15252/embj.2022112012. Epub 2022 Oct 10. EMBO J. 2022. PMID: 36215692 Free PMC article.
-
Garcinol-A Natural Histone Acetyltransferase Inhibitor and New Anti-Cancer Epigenetic Drug.Int J Mol Sci. 2021 Mar 11;22(6):2828. doi: 10.3390/ijms22062828. Int J Mol Sci. 2021. PMID: 33799504 Free PMC article. Review.
-
The role of protein acetylation in carcinogenesis and targeted drug discovery.Front Endocrinol (Lausanne). 2022 Sep 12;13:972312. doi: 10.3389/fendo.2022.972312. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36171897 Free PMC article. Review.
-
Small-molecule CBP/p300 histone acetyltransferase inhibition mobilizes leukocytes from the bone marrow via the endocrine stress response.Immunity. 2024 Feb 13;57(2):364-378.e9. doi: 10.1016/j.immuni.2024.01.005. Epub 2024 Jan 31. Immunity. 2024. PMID: 38301651 Free PMC article.
-
Nutritional Epigenetics in Cancer.Adv Nutr. 2022 Oct 2;13(5):1748-1761. doi: 10.1093/advances/nmac039. Adv Nutr. 2022. PMID: 35421212 Free PMC article. Review.
References
-
- Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell. Biol. 2014;15:703–708. - PubMed
-
- Simon RP, Robaa D, Alhalabi Z, Sippl W, Jung M. KATching-Up on Small Molecule Modulators of Lysine Acetyltransferases. J. Med. Chem. 2016;59:1249–1270. - PubMed
-
- Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225–4231. - PubMed
-
- Balasubramanyam K, et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J. Biol. Chem. 2004;279:33716–33726. - PubMed
-
- Lau OD, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell. 2000;5:589–595. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
