

Variant Interpretation: Functional Assays to the Rescue
- PMID: 28886340
- PMCID: PMC5590843
- DOI: 10.1016/j.ajhg.2017.07.014
Variant Interpretation: Functional Assays to the Rescue
Abstract
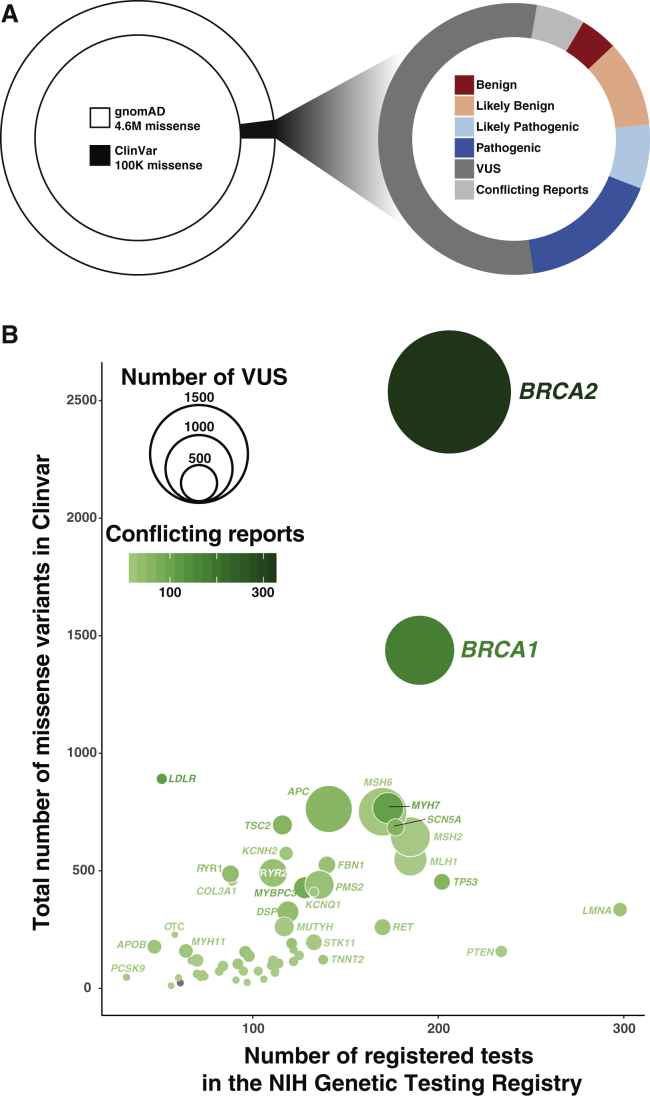
Classical genetic approaches for interpreting variants, such as case-control or co-segregation studies, require finding many individuals with each variant. Because the overwhelming majority of variants are present in only a few living humans, this strategy has clear limits. Fully realizing the clinical potential of genetics requires that we accurately infer pathogenicity even for rare or private variation. Many computational approaches to predicting variant effects have been developed, but they can identify only a small fraction of pathogenic variants with the high confidence that is required in the clinic. Experimentally measuring a variant's functional consequences can provide clearer guidance, but individual assays performed only after the discovery of the variant are both time and resource intensive. Here, we discuss how multiplex assays of variant effect (MAVEs) can be used to measure the functional consequences of all possible variants in disease-relevant loci for a variety of molecular and cellular phenotypes. The resulting large-scale functional data can be combined with machine learning and clinical knowledge for the development of "lookup tables" of accurate pathogenicity predictions. A coordinated effort to produce, analyze, and disseminate large-scale functional data generated by multiplex assays could be essential to addressing the variant-interpretation crisis.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
Multi-objective prioritization of genes for high-throughput functional assays towards improved clinical variant classification.Pac Symp Biocomput. 2023;28:323-334. Pac Symp Biocomput. 2023. PMID: 36540988 Free PMC article.
-
ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants.Am J Hum Genet. 2018 Oct 4;103(4):474-483. doi: 10.1016/j.ajhg.2018.08.005. Epub 2018 Sep 13. Am J Hum Genet. 2018. PMID: 30220433 Free PMC article.
-
Multiplexed assays of variant effects contribute to a growing genotype-phenotype atlas.Hum Genet. 2018 Sep;137(9):665-678. doi: 10.1007/s00439-018-1916-x. Epub 2018 Aug 2. Hum Genet. 2018. PMID: 30073413 Free PMC article. Review.
-
Disease variant prediction with deep generative models of evolutionary data.Nature. 2021 Nov;599(7883):91-95. doi: 10.1038/s41586-021-04043-8. Epub 2021 Oct 27. Nature. 2021. PMID: 34707284
-
From variant to function in human disease genetics.Science. 2021 Sep 24;373(6562):1464-1468. doi: 10.1126/science.abi8207. Epub 2021 Sep 23. Science. 2021. PMID: 34554789 Review.
Cited by
-
Accurate prediction of functional effect of single amino acid variants with deep learning.Comput Struct Biotechnol J. 2023 Nov 10;21:5776-5784. doi: 10.1016/j.csbj.2023.11.017. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 38074467 Free PMC article.
-
Intragenic compensation through the lens of deep mutational scanning.Biophys Rev. 2022 Oct 26;14(5):1161-1182. doi: 10.1007/s12551-022-01005-w. eCollection 2022 Oct. Biophys Rev. 2022. PMID: 36345285 Free PMC article. Review.
-
Comprehensive characterization of amino acid positions in protein structures reveals molecular effect of missense variants.Proc Natl Acad Sci U S A. 2020 Nov 10;117(45):28201-28211. doi: 10.1073/pnas.2002660117. Epub 2020 Oct 26. Proc Natl Acad Sci U S A. 2020. PMID: 33106425 Free PMC article.
-
Recommendations for the collection and use of multiplexed functional data for clinical variant interpretation.Genome Med. 2019 Dec 20;11(1):85. doi: 10.1186/s13073-019-0698-7. Genome Med. 2019. PMID: 31862013 Free PMC article.
-
Computational and experimental methods for classifying variants of unknown clinical significance.Cold Spring Harb Mol Case Stud. 2022 Apr 28;8(3):a006196. doi: 10.1101/mcs.a006196. Print 2022 Apr. Cold Spring Harb Mol Case Stud. 2022. PMID: 35483875 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
