

Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology
- PMID: 28874689
- PMCID: PMC5585393
- DOI: 10.1038/s41598-017-09654-8
Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology
Abstract
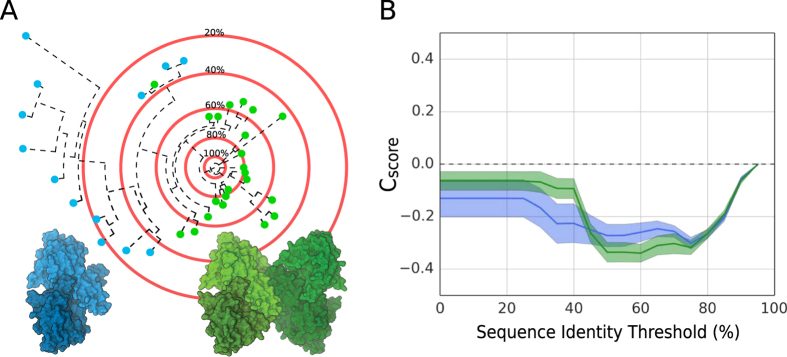
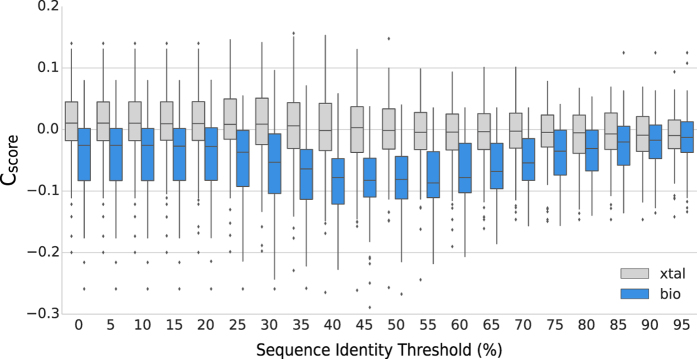
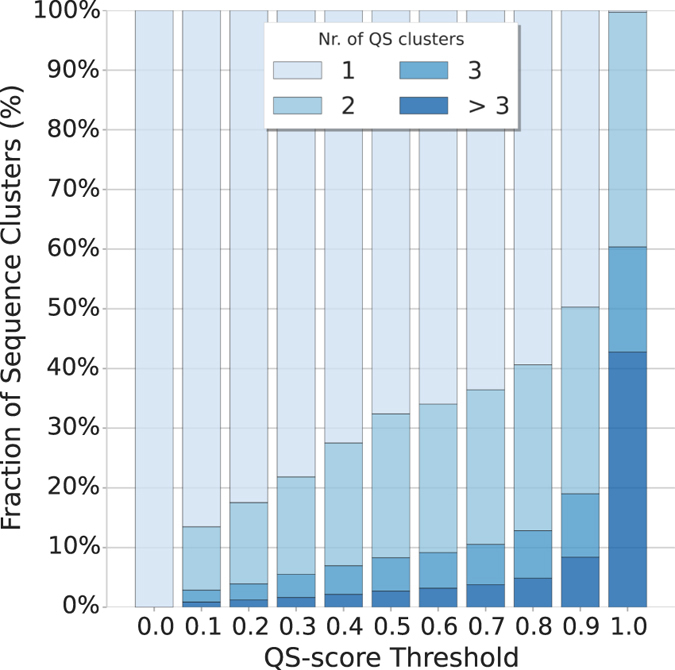
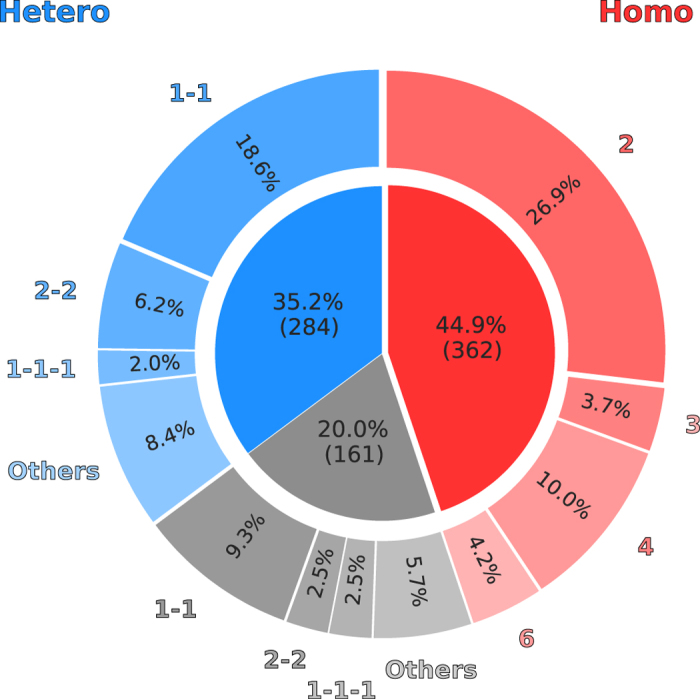
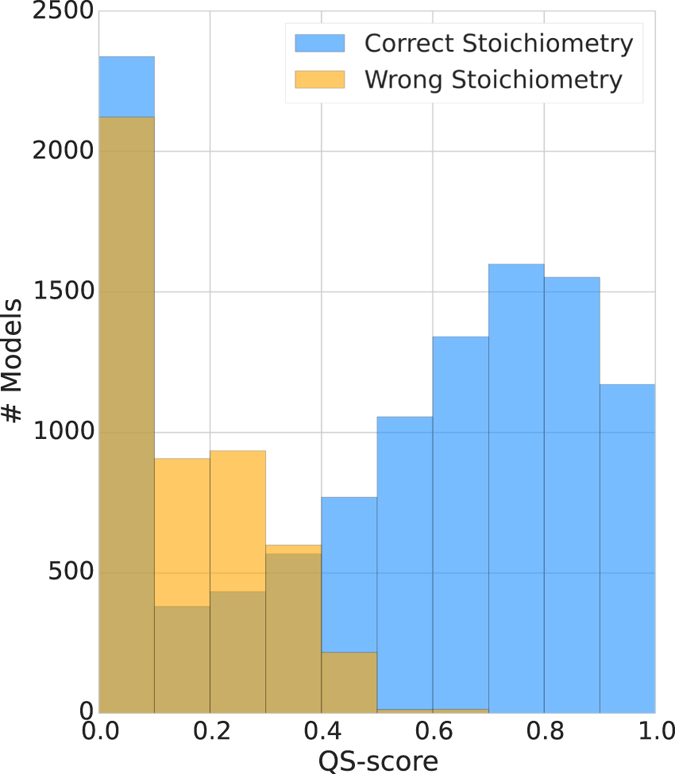
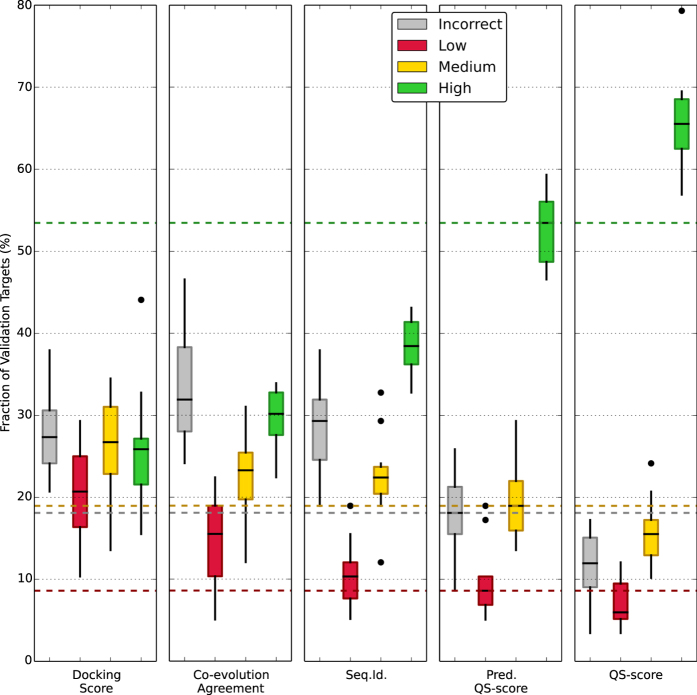
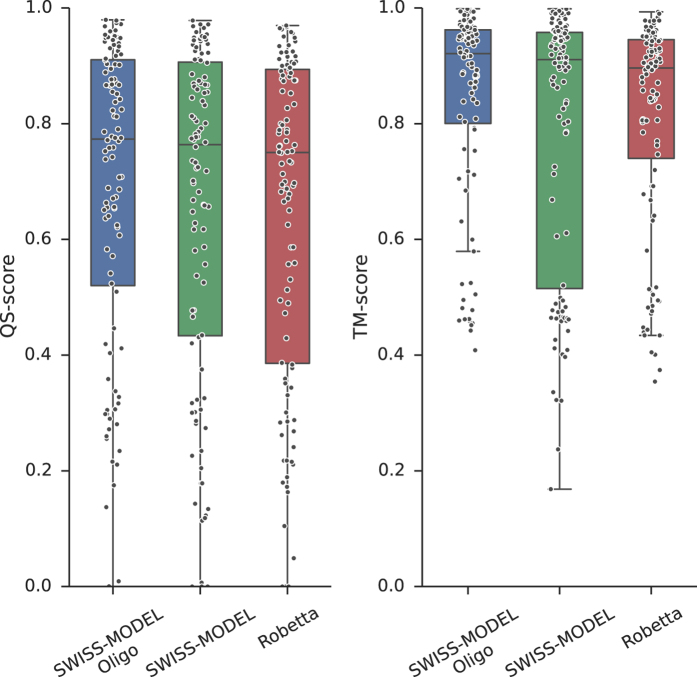
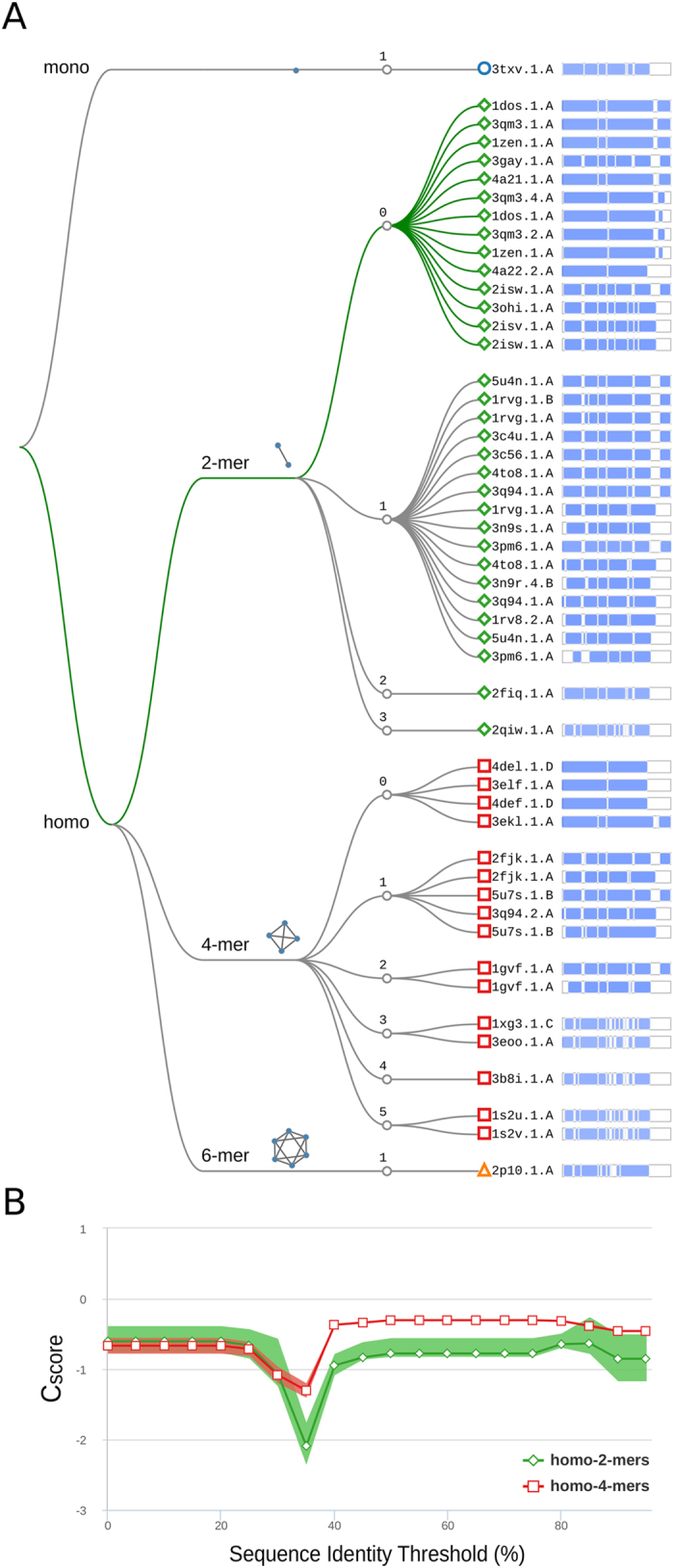
Cellular processes often depend on interactions between proteins and the formation of macromolecular complexes. The impairment of such interactions can lead to deregulation of pathways resulting in disease states, and it is hence crucial to gain insights into the nature of macromolecular assemblies. Detailed structural knowledge about complexes and protein-protein interactions is growing, but experimentally determined three-dimensional multimeric assemblies are outnumbered by complexes supported by non-structural experimental evidence. Here, we aim to fill this gap by modeling multimeric structures by homology, only using amino acid sequences to infer the stoichiometry and the overall structure of the assembly. We ask which properties of proteins within a family can assist in the prediction of correct quaternary structure. Specifically, we introduce a description of protein-protein interface conservation as a function of evolutionary distance to reduce the noise in deep multiple sequence alignments. We also define a distance measure to structurally compare homologous multimeric protein complexes. This allows us to hierarchically cluster protein structures and quantify the diversity of alternative biological assemblies known today. We find that a combination of conservation scores, structural clustering, and classical interface descriptors, can improve the selection of homologous protein templates leading to reliable models of protein complexes.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Structural and functional specificity of small heat shock protein HspB1 and HspB4, two cellular partners of HspB5: role of the in vitro hetero-complex formation in chaperone activity.Biochimie. 2012 Apr;94(4):975-84. doi: 10.1016/j.biochi.2011.12.018. Epub 2011 Dec 26. Biochimie. 2012. PMID: 22210387
-
Comparative analyses of quaternary arrangements in homo-oligomeric proteins in superfamilies: Functional implications.Proteins. 2016 Sep;84(9):1190-202. doi: 10.1002/prot.25065. Epub 2016 Jun 8. Proteins. 2016. PMID: 27177429
-
Modeling of protein complexes in CAPRI Round 37 using template-based approach combined with model selection.Proteins. 2018 Mar;86 Suppl 1:292-301. doi: 10.1002/prot.25378. Epub 2017 Sep 23. Proteins. 2018. PMID: 28905467
-
Protein modeling: what happened to the "protein structure gap"?Structure. 2013 Sep 3;21(9):1531-40. doi: 10.1016/j.str.2013.08.007. Structure. 2013. PMID: 24010712 Free PMC article. Review.
-
Pharmacological interference with protein-protein interactions mediated by coiled-coil motifs.Handb Exp Pharmacol. 2008;(186):461-82. doi: 10.1007/978-3-540-72843-6_19. Handb Exp Pharmacol. 2008. PMID: 18491064 Review.
Cited by
-
Structural characterization of protective non-neutralizing antibodies targeting Crimean-Congo hemorrhagic fever virus.Nat Commun. 2022 Nov 26;13(1):7298. doi: 10.1038/s41467-022-34923-0. Nat Commun. 2022. PMID: 36435827 Free PMC article.
-
Underpinning the molecular programming attributing heat stress associated thermotolerance in tea (Camellia sinensis (L.) O. Kuntze).Hortic Res. 2021 May 1;8(1):99. doi: 10.1038/s41438-021-00532-z. Hortic Res. 2021. PMID: 33931616 Free PMC article.
-
The molecular basis for sarcomere organization in vertebrate skeletal muscle.Cell. 2021 Apr 15;184(8):2135-2150.e13. doi: 10.1016/j.cell.2021.02.047. Epub 2021 Mar 24. Cell. 2021. PMID: 33765442 Free PMC article.
-
Common polymorphic OTC variants can act as genetic modifiers of enzymatic activity.Hum Mutat. 2021 Aug;42(8):978-989. doi: 10.1002/humu.24221. Epub 2021 Jun 3. Hum Mutat. 2021. PMID: 34015158 Free PMC article.
-
Melastatin subfamily Transient Receptor Potential channels support spermatogenesis in planarian flatworms.bioRxiv [Preprint]. 2024 Sep 3:2024.09.01.610670. doi: 10.1101/2024.09.01.610670. bioRxiv. 2024. PMID: 39282438 Free PMC article. Preprint.
References
-
- Terradot L, et al. Biochemical characterization of protein complexes from the Helicobacter pylori protein interaction map: strategies for complex formation and evidence for novel interactions within type IV secretion systems. Mol Cell Proteomics. 2004;3:809–819. doi: 10.1074/mcp.M400048-MCP200. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
