

The TRIMendous Role of TRIMs in Virus-Host Interactions
- PMID: 28829373
- PMCID: PMC5620554
- DOI: 10.3390/vaccines5030023
The TRIMendous Role of TRIMs in Virus-Host Interactions
Abstract
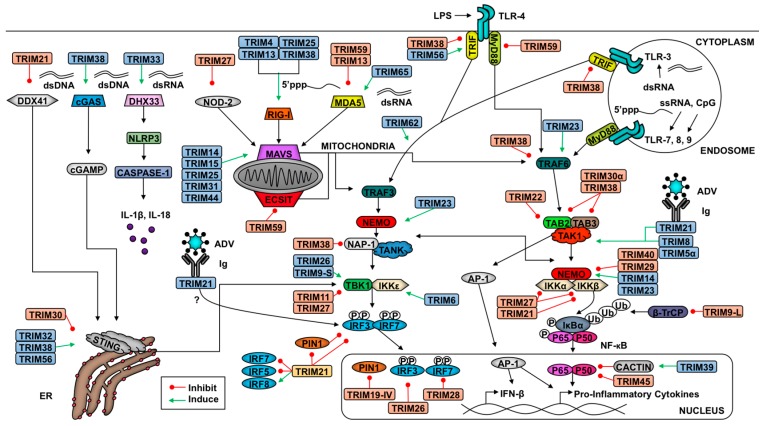
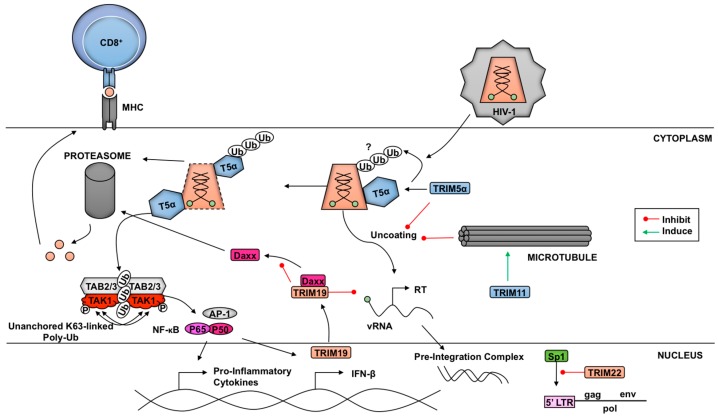
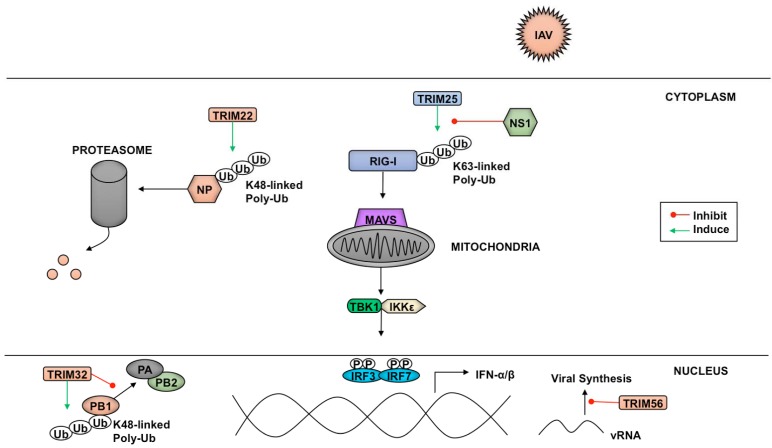
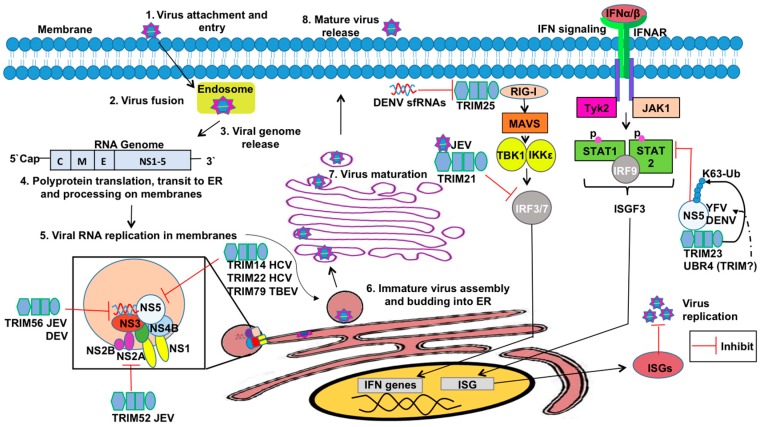
The innate antiviral response is integral in protecting the host against virus infection. Many proteins regulate these signaling pathways including ubiquitin enzymes. The ubiquitin-activating (E1), -conjugating (E2), and -ligating (E3) enzymes work together to link ubiquitin, a small protein, onto other ubiquitin molecules or target proteins to mediate various effector functions. The tripartite motif (TRIM) protein family is a group of E3 ligases implicated in the regulation of a variety of cellular functions including cell cycle progression, autophagy, and innate immunity. Many antiviral signaling pathways, including type-I interferon and NF-κB, are TRIM-regulated, thus influencing the course of infection. Additionally, several TRIMs directly restrict viral replication either through proteasome-mediated degradation of viral proteins or by interfering with different steps of the viral replication cycle. In addition, new studies suggest that TRIMs can exert their effector functions via the synthesis of unconventional polyubiquitin chains, including unanchored (non-covalently attached) polyubiquitin chains. TRIM-conferred viral inhibition has selected for viruses that encode direct and indirect TRIM antagonists. Furthermore, new evidence suggests that the same antagonists encoded by viruses may hijack TRIM proteins to directly promote virus replication. Here, we describe numerous virus-TRIM interactions and novel roles of TRIMs during virus infections.
Keywords: E3-ubiquitin ligase; innate immunity; tripartite motif (TRIM); type-I interferons; ubiquitin; unanchored polyubiquitin; viral antagonism; virus infection.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
To TRIM or not to TRIM: the balance of host-virus interactions mediated by the ubiquitin system.J Gen Virol. 2019 Dec;100(12):1641-1662. doi: 10.1099/jgv.0.001341. J Gen Virol. 2019. PMID: 31661051 Free PMC article.
-
TRIM Proteins in Host Defense and Viral Pathogenesis.Curr Clin Microbiol Rep. 2020;7(4):101-114. doi: 10.1007/s40588-020-00150-8. Epub 2020 Aug 8. Curr Clin Microbiol Rep. 2020. PMID: 32837832 Free PMC article. Review.
-
The Roles of TRIMs in Antiviral Innate Immune Signaling.Front Cell Infect Microbiol. 2021 Mar 15;11:628275. doi: 10.3389/fcimb.2021.628275. eCollection 2021. Front Cell Infect Microbiol. 2021. PMID: 33791238 Free PMC article. Review.
-
TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity.J Mol Biol. 2014 Mar 20;426(6):1265-84. doi: 10.1016/j.jmb.2013.12.005. Epub 2013 Dec 12. J Mol Biol. 2014. PMID: 24333484 Free PMC article. Review.
-
The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication.J Virol. 2017 Aug 24;91(18):e00833-17. doi: 10.1128/JVI.00833-17. Print 2017 Sep 15. J Virol. 2017. PMID: 28679761 Free PMC article.
Cited by
-
Crystal structure and mutational analysis of the human TRIM7 B30.2 domain provide insights into the molecular basis of its binding to glycogenin-1.J Biol Chem. 2021 Jan-Jun;296:100772. doi: 10.1016/j.jbc.2021.100772. Epub 2021 May 11. J Biol Chem. 2021. PMID: 33989636 Free PMC article.
-
Unconventional RNA-binding proteins step into the virus-host battlefront.Wiley Interdiscip Rev RNA. 2018 Nov;9(6):e1498. doi: 10.1002/wrna.1498. Epub 2018 Aug 9. Wiley Interdiscip Rev RNA. 2018. PMID: 30091184 Free PMC article. Review.
-
Regulation of Tripartite Motif-Containing Proteins on Immune Response and Viral Evasion.Front Microbiol. 2021 Dec 1;12:794882. doi: 10.3389/fmicb.2021.794882. eCollection 2021. Front Microbiol. 2021. PMID: 34925304 Free PMC article. Review.
-
RIG-I-like receptors: their regulation and roles in RNA sensing.Nat Rev Immunol. 2020 Sep;20(9):537-551. doi: 10.1038/s41577-020-0288-3. Epub 2020 Mar 13. Nat Rev Immunol. 2020. PMID: 32203325 Free PMC article. Review.
-
Identification of Interleukin1β as an Amplifier of Interferon alpha-induced Antiviral Responses.PLoS Pathog. 2020 Oct 1;16(10):e1008461. doi: 10.1371/journal.ppat.1008461. eCollection 2020 Oct. PLoS Pathog. 2020. PMID: 33002089 Free PMC article.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
