

Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation
- PMID: 28825701
- PMCID: PMC5605457
- DOI: 10.1038/ni.3818
Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation
Abstract
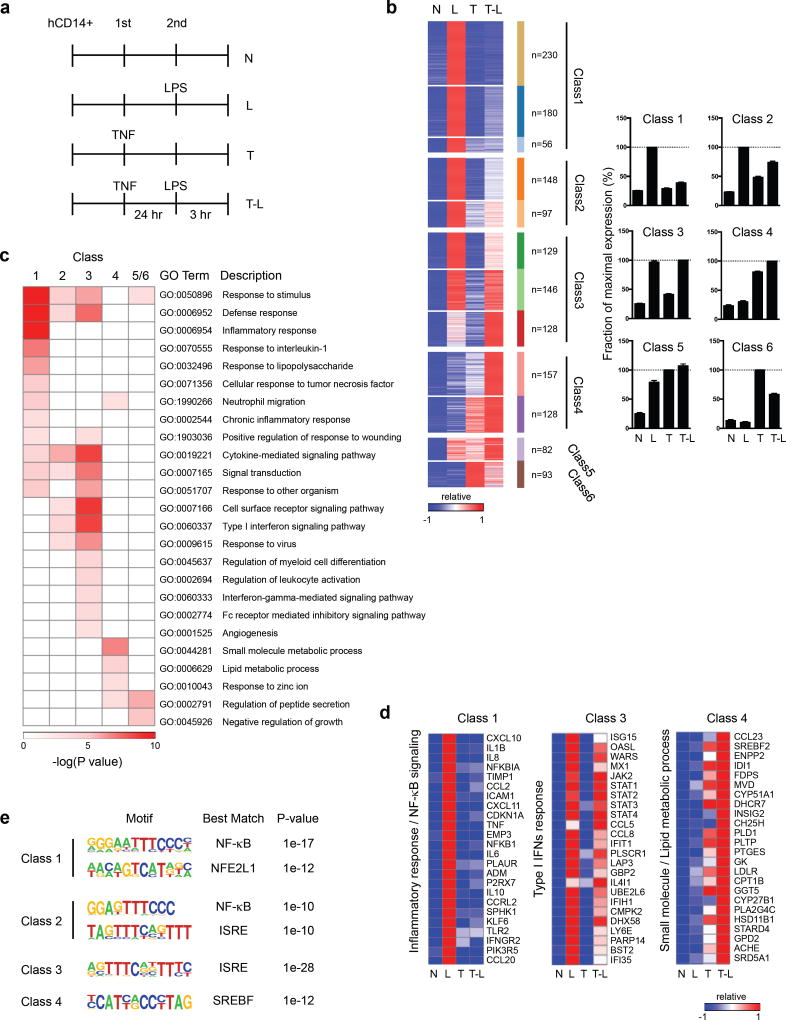
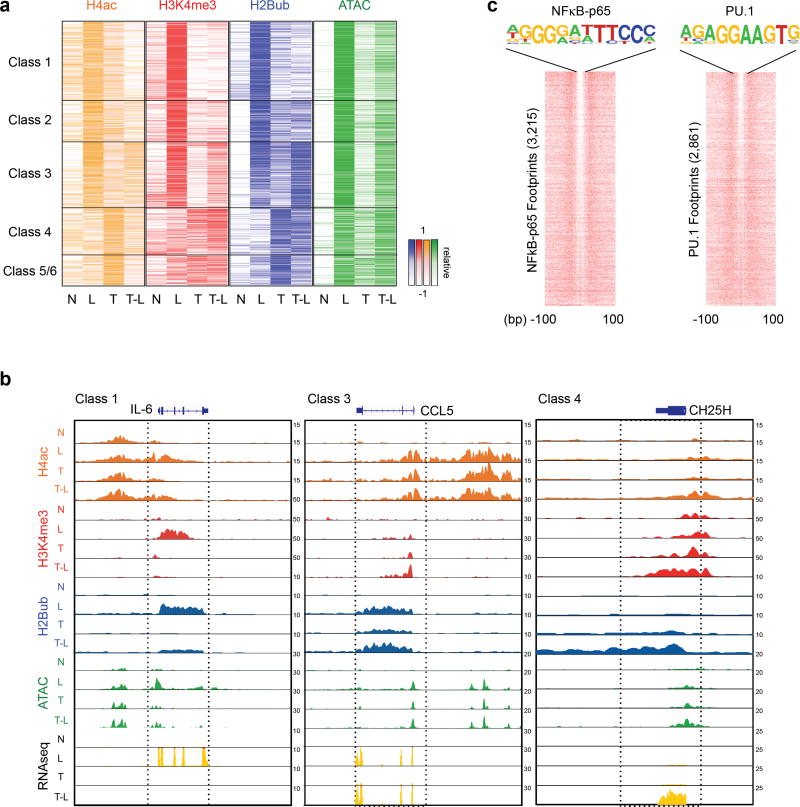
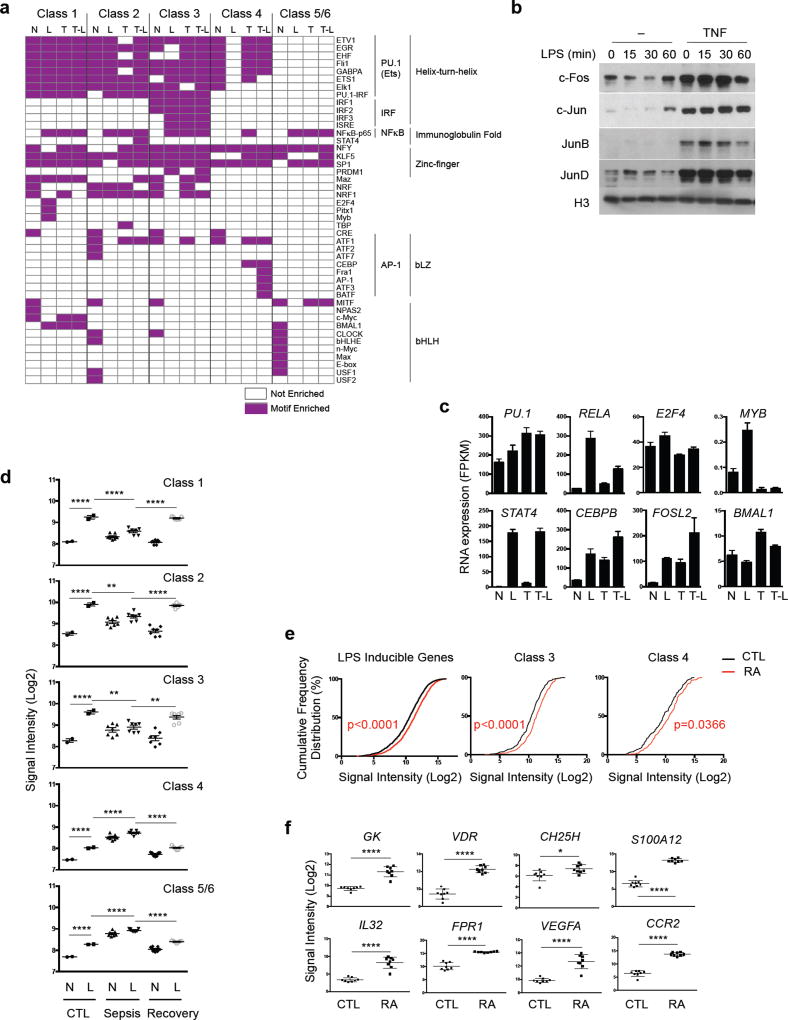
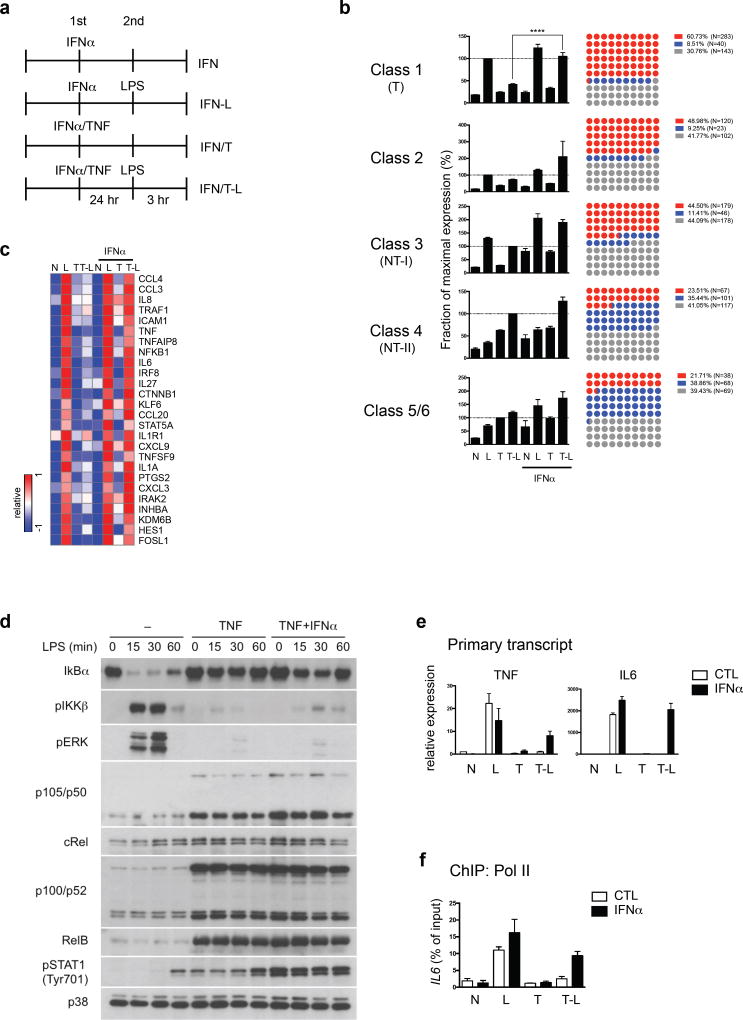
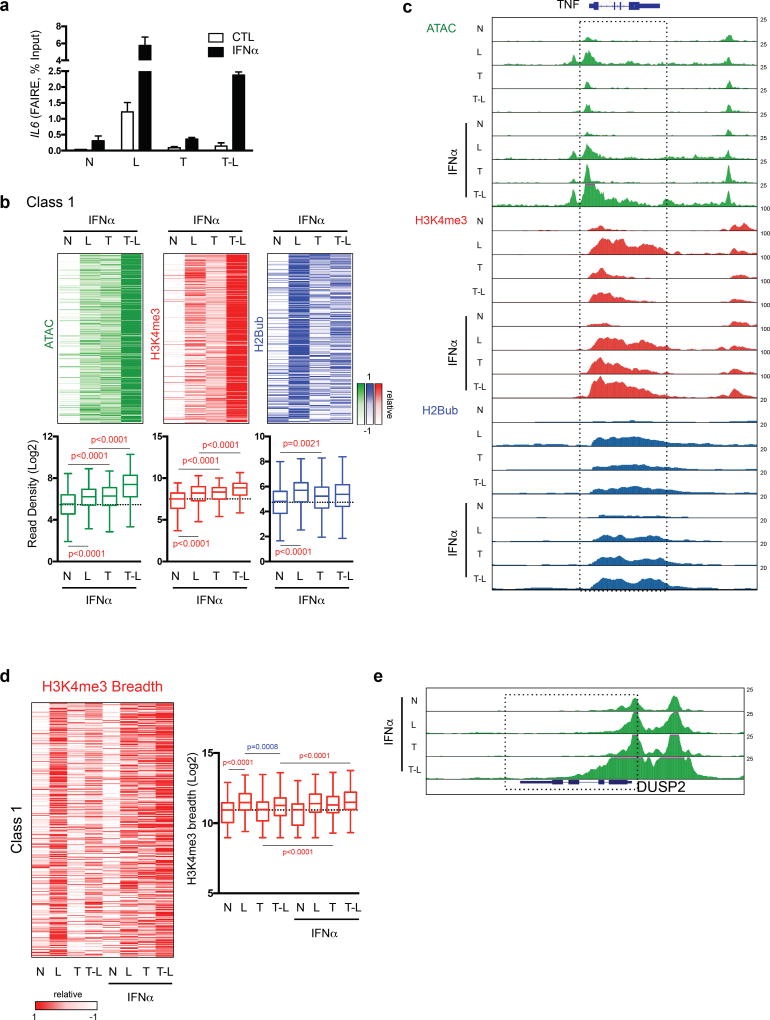
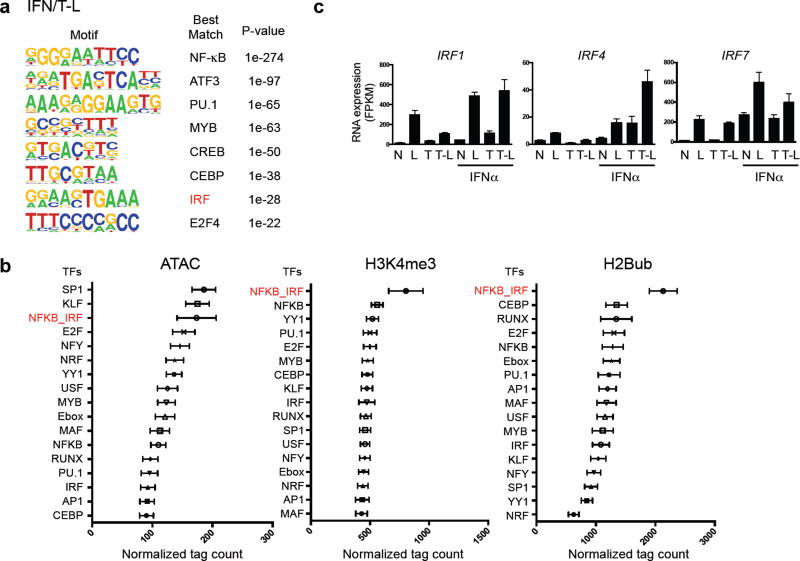
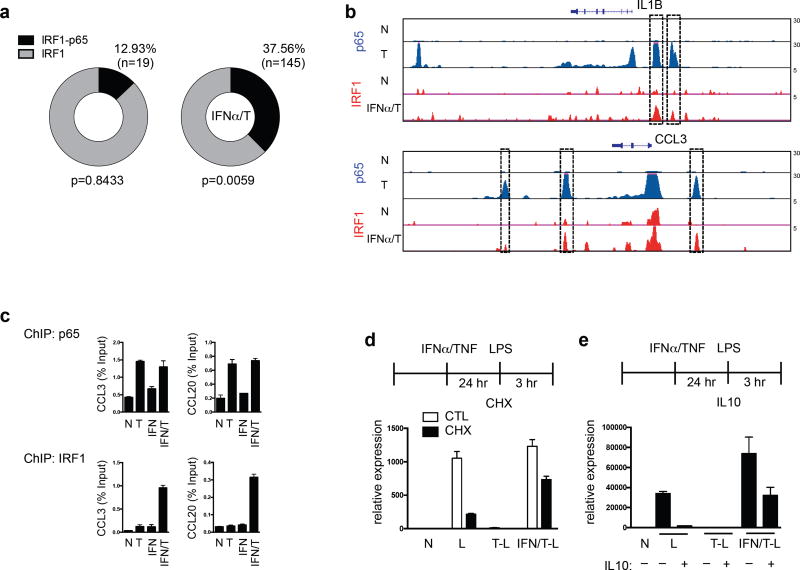
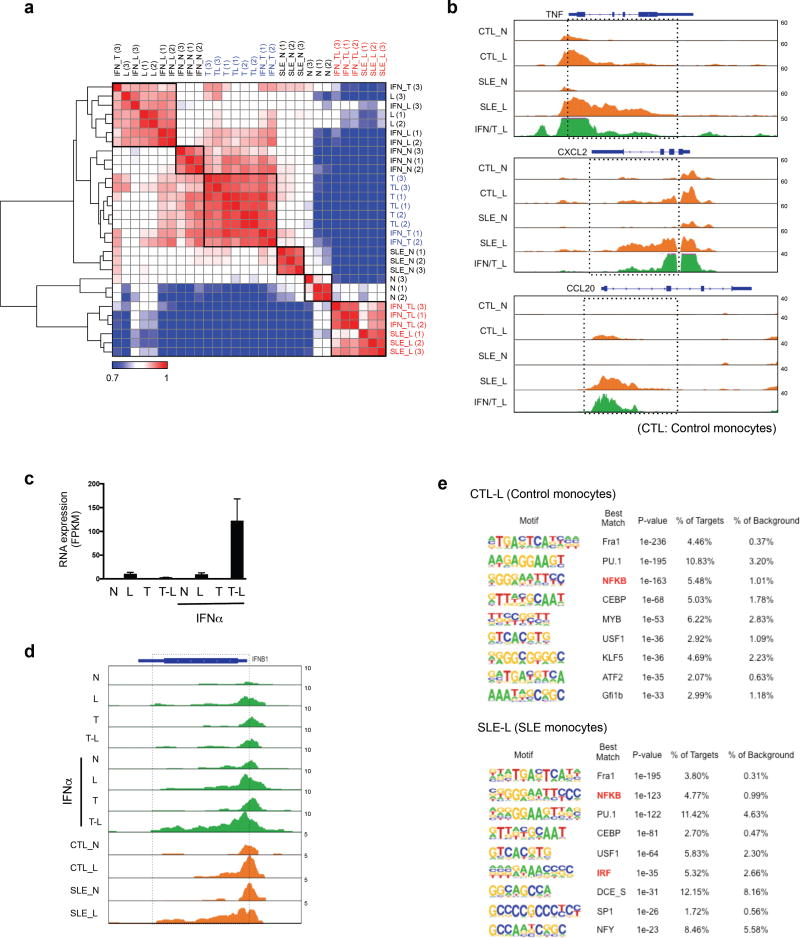
Cross-regulation of Toll-like receptor (TLR) responses by cytokines is essential for effective host defense, avoidance of toxicity and homeostasis, but the underlying mechanisms are not well understood. Our comprehensive epigenomics approach to the analysis of human macrophages showed that the proinflammatory cytokines TNF and type I interferons induced transcriptional cascades that altered chromatin states to broadly reprogram responses induced by TLR4. TNF tolerized genes encoding inflammatory molecules to prevent toxicity while preserving the induction of genes encoding antiviral and metabolic molecules. Type I interferons potentiated the inflammatory function of TNF by priming chromatin to prevent the silencing of target genes of the transcription factor NF-κB that encode inflammatory molecules. The priming of chromatin enabled robust transcriptional responses to weak upstream signals. Similar chromatin regulation occurred in human diseases. Our findings reveal that signaling crosstalk between interferons and TNF is integrated at the level of chromatin to reprogram inflammatory responses, and identify previously unknown functions and mechanisms of action of these cytokines.
Figures
Comment in
-
Inflammation: Cytokines alter inflammatory responses via chromatin changes.Nat Rev Rheumatol. 2017 Sep 22;13(10):569. doi: 10.1038/nrrheum.2017.154. Nat Rev Rheumatol. 2017. PMID: 28935944 No abstract available.
-
The yin and yang of cytokine priming on the macrophage epigenome.Sci Immunol. 2017 Nov 3;2(17):eaaq0016. doi: 10.1126/sciimmunol.aaq0016. Sci Immunol. 2017. PMID: 29101211
Similar articles
-
Role of CD44 in Regulating TLR2 Activation of Human Macrophages and Downstream Expression of Proinflammatory Cytokines.J Immunol. 2018 Jan 15;200(2):758-767. doi: 10.4049/jimmunol.1700713. Epub 2017 Dec 1. J Immunol. 2018. PMID: 29196459 Free PMC article.
-
A novel crosstalk between TLR4- and NOD2-mediated signaling in the regulation of intestinal inflammation.Sci Rep. 2015 Jul 8;5:12018. doi: 10.1038/srep12018. Sci Rep. 2015. PMID: 26153766 Free PMC article.
-
A TNF-induced gene expression program under oscillatory NF-kappaB control.BMC Genomics. 2005 Sep 28;6:137. doi: 10.1186/1471-2164-6-137. BMC Genomics. 2005. PMID: 16191192 Free PMC article.
-
Epigenetic Mechanisms Governing Innate Inflammatory Responses.J Interferon Cytokine Res. 2016 Jul;36(7):454-61. doi: 10.1089/jir.2016.0003. J Interferon Cytokine Res. 2016. PMID: 27379867 Free PMC article. Review.
-
Interferon target-gene expression and epigenomic signatures in health and disease.Nat Immunol. 2019 Dec;20(12):1574-1583. doi: 10.1038/s41590-019-0466-2. Epub 2019 Nov 19. Nat Immunol. 2019. PMID: 31745335 Free PMC article. Review.
Cited by
-
Teaching Old Dogs New Tricks? The Plasticity of Lung Alveolar Macrophage Subsets.Trends Immunol. 2020 Oct;41(10):864-877. doi: 10.1016/j.it.2020.08.008. Epub 2020 Sep 4. Trends Immunol. 2020. PMID: 32896485 Free PMC article. Review.
-
Sensing of SARS-CoV-2 by pDCs and their subsequent production of IFN-I contribute to macrophage-induced cytokine storm during COVID-19.Sci Immunol. 2022 Sep 9;7(75):eadd4906. doi: 10.1126/sciimmunol.add4906. Epub 2022 Sep 9. Sci Immunol. 2022. PMID: 36083891 Free PMC article.
-
Lack of Ikaros Deregulates Inflammatory Gene Programs in T Cells.J Immunol. 2019 Feb 15;202(4):1112-1123. doi: 10.4049/jimmunol.1801270. Epub 2019 Jan 11. J Immunol. 2019. PMID: 30635395 Free PMC article.
-
Trabectedin Reveals a Strategy of Immunomodulation in Chronic Lymphocytic Leukemia.Cancer Immunol Res. 2019 Dec;7(12):2036-2051. doi: 10.1158/2326-6066.CIR-19-0152. Epub 2019 Sep 17. Cancer Immunol Res. 2019. PMID: 31530560 Free PMC article.
-
Two Faces of Macrophages: Training and Tolerance.Biomedicines. 2021 Nov 2;9(11):1596. doi: 10.3390/biomedicines9111596. Biomedicines. 2021. PMID: 34829825 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
