

De novo mutations in inhibitors of Wnt, BMP, and Ras/ERK signaling pathways in non-syndromic midline craniosynostosis
- PMID: 28808027
- PMCID: PMC5584457
- DOI: 10.1073/pnas.1709255114
De novo mutations in inhibitors of Wnt, BMP, and Ras/ERK signaling pathways in non-syndromic midline craniosynostosis
Abstract
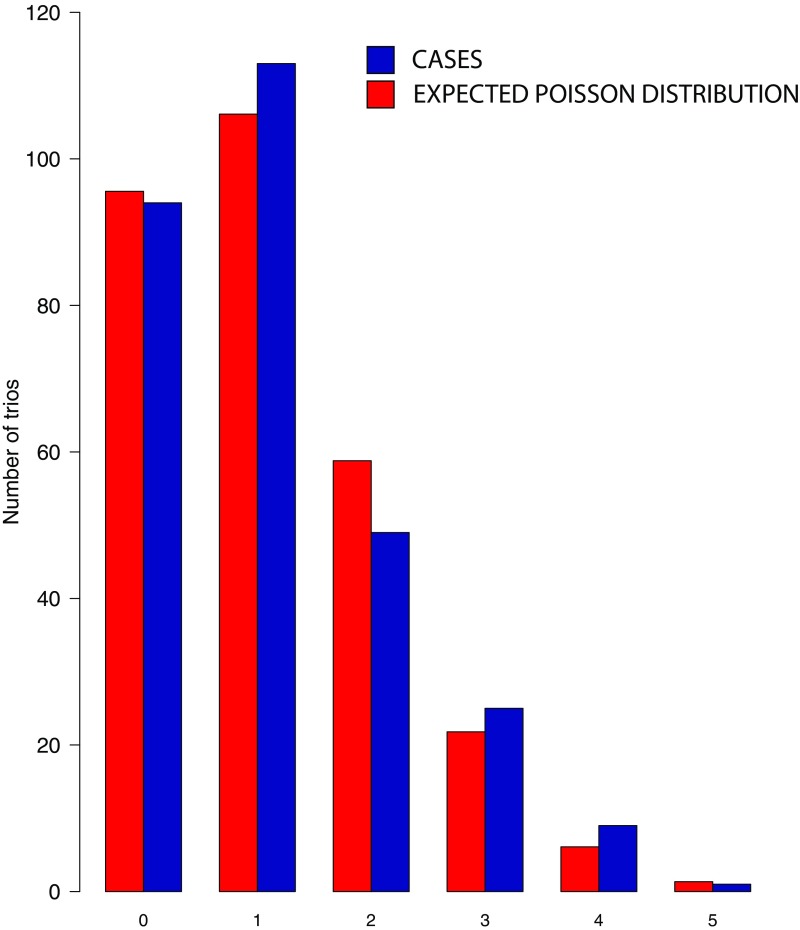
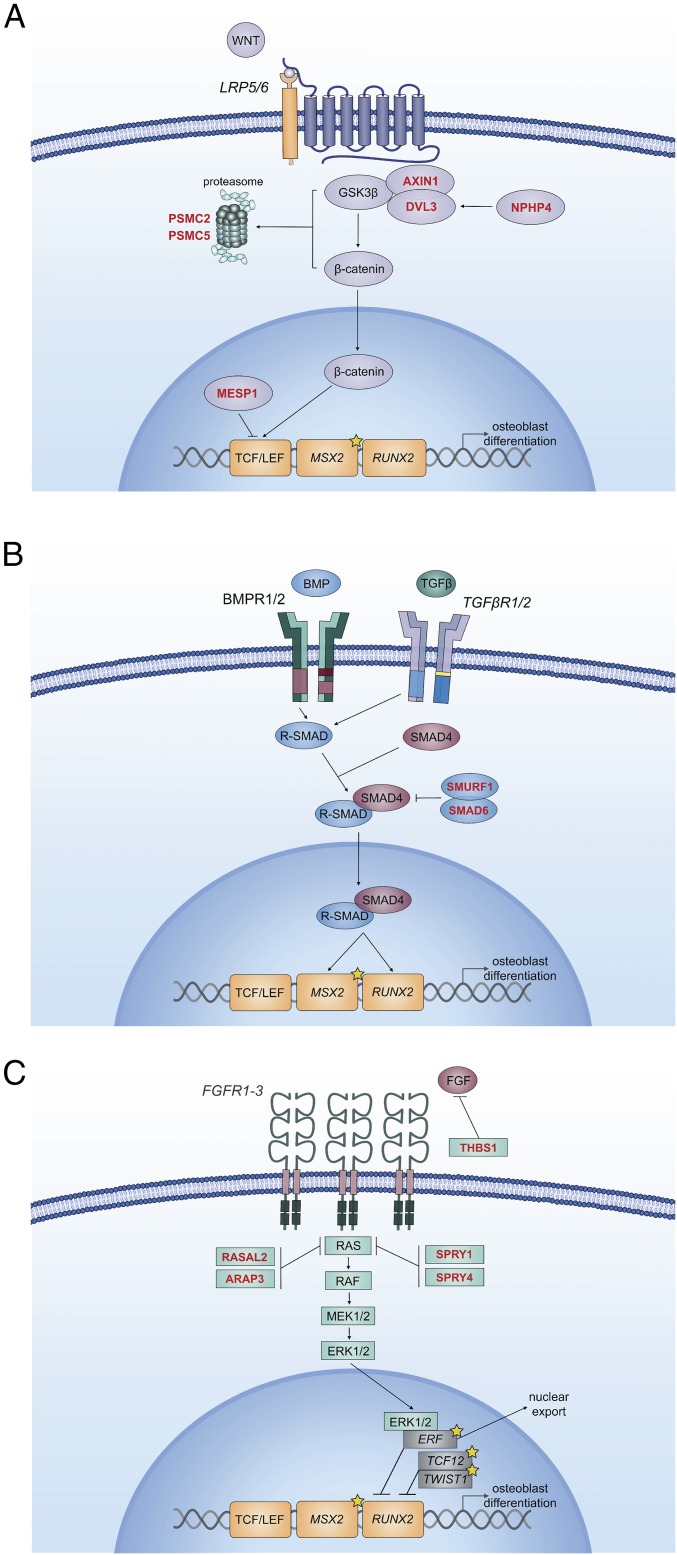
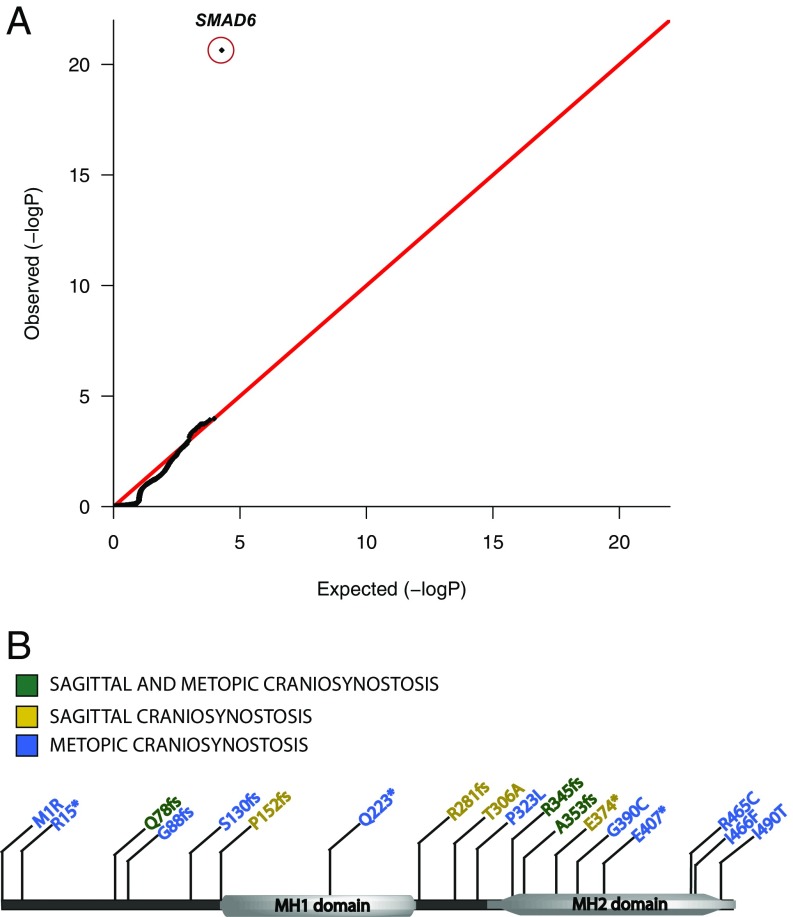
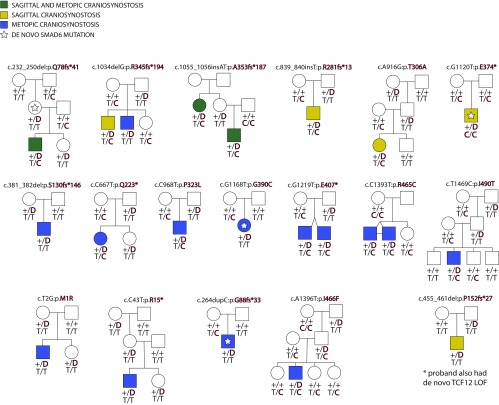
Non-syndromic craniosynostosis (NSC) is a frequent congenital malformation in which one or more cranial sutures fuse prematurely. Mutations causing rare syndromic craniosynostoses in humans and engineered mouse models commonly increase signaling of the Wnt, bone morphogenetic protein (BMP), or Ras/ERK pathways, converging on shared nuclear targets that promote bone formation. In contrast, the genetics of NSC is largely unexplored. More than 95% of NSC is sporadic, suggesting a role for de novo mutations. Exome sequencing of 291 parent-offspring trios with midline NSC revealed 15 probands with heterozygous damaging de novo mutations in 12 negative regulators of Wnt, BMP, and Ras/ERK signaling (10.9-fold enrichment, P = 2.4 × 10-11). SMAD6 had 4 de novo and 14 transmitted mutations; no other gene had more than 1. Four familial NSC kindreds had mutations in genes previously implicated in syndromic disease. Collectively, these mutations contribute to 10% of probands. Mutations are predominantly loss-of-function, implicating haploinsufficiency as a frequent mechanism. A common risk variant near BMP2 increased the penetrance of SMAD6 mutations and was overtransmitted to patients with de novo mutations in other genes in these pathways, supporting a frequent two-locus pathogenesis. These findings implicate new genes in NSC and demonstrate related pathophysiology of common non-syndromic and rare syndromic craniosynostoses. These findings have implications for diagnosis, risk of recurrence, and risk of adverse neurodevelopmental outcomes. Finally, the use of pathways identified in rare syndromic disease to find genes accounting for non-syndromic cases may prove broadly relevant to understanding other congenital disorders featuring high locus heterogeneity.
Keywords: BMP signaling; Ras/ERK signaling; Wnt signaling; craniosynostosis; de novo mutation.
Conflict of interest statement
Conflict of interest statement: J.S. and R.P.L. were among 42 coauthors on a 2015 review article.
Figures
Similar articles
-
Signaling Mechanisms Underlying Genetic Pathophysiology of Craniosynostosis.Int J Biol Sci. 2019 Jan 1;15(2):298-311. doi: 10.7150/ijbs.29183. eCollection 2019. Int J Biol Sci. 2019. PMID: 30745822 Free PMC article. Review.
-
Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles.Elife. 2016 Sep 8;5:e20125. doi: 10.7554/eLife.20125. Elife. 2016. PMID: 27606499 Free PMC article.
-
Mutations in TFAP2B and previously unimplicated genes of the BMP, Wnt, and Hedgehog pathways in syndromic craniosynostosis.Proc Natl Acad Sci U S A. 2019 Jul 23;116(30):15116-15121. doi: 10.1073/pnas.1902041116. Epub 2019 Jul 10. Proc Natl Acad Sci U S A. 2019. PMID: 31292255 Free PMC article.
-
De novo mutations in the BMP signaling pathway in lambdoid craniosynostosis.Hum Genet. 2023 Jan;142(1):21-32. doi: 10.1007/s00439-022-02477-2. Epub 2022 Aug 23. Hum Genet. 2023. PMID: 35997807
-
Craniosynostosis and Noonan syndrome with KRAS mutations: Expanding the phenotype with a case report and review of the literature.Am J Med Genet A. 2015 Nov;167A(11):2657-63. doi: 10.1002/ajmg.a.37259. Epub 2015 Aug 6. Am J Med Genet A. 2015. PMID: 26249544 Review.
Cited by
-
Craniosynostosis, inner ear, and renal anomalies in a child with complete loss of SPRY1 (sprouty homolog 1) function.J Med Genet. 2023 Jul;60(7):712-716. doi: 10.1136/jmg-2022-108946. Epub 2022 Dec 21. J Med Genet. 2023. PMID: 36543535 Free PMC article.
-
Proteomic Analysis of Exosomes during Cardiogenic Differentiation of Human Pluripotent Stem Cells.Cells. 2021 Oct 1;10(10):2622. doi: 10.3390/cells10102622. Cells. 2021. PMID: 34685602 Free PMC article.
-
Dysregulation of BMP, Wnt, and Insulin Signaling in Fragile X Syndrome.Front Cell Dev Biol. 2022 Jul 6;10:934662. doi: 10.3389/fcell.2022.934662. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35880195 Free PMC article. Review.
-
Signaling Mechanisms Underlying Genetic Pathophysiology of Craniosynostosis.Int J Biol Sci. 2019 Jan 1;15(2):298-311. doi: 10.7150/ijbs.29183. eCollection 2019. Int J Biol Sci. 2019. PMID: 30745822 Free PMC article. Review.
-
Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy.Eur J Hum Genet. 2019 Jul;27(7):1044-1053. doi: 10.1038/s41431-019-0363-z. Epub 2019 Feb 22. Eur J Hum Genet. 2019. PMID: 30796334 Free PMC article. Clinical Trial.
References
-
- Slater BJ, et al. Cranial sutures: A brief review. Plast Reconstr Surg. 2008;121:170e–178e. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
