

Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo
- PMID: 28737770
- PMCID: PMC5540176
- DOI: 10.1038/ncb3573
Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo
Abstract
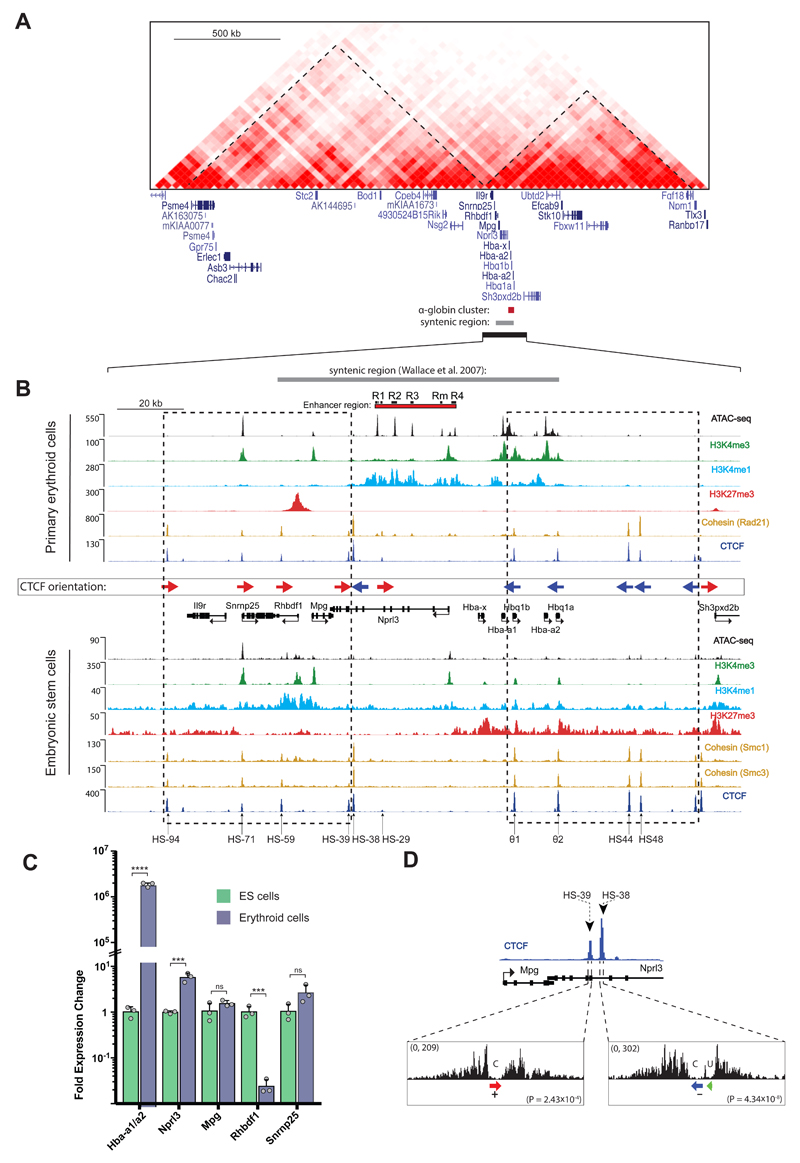
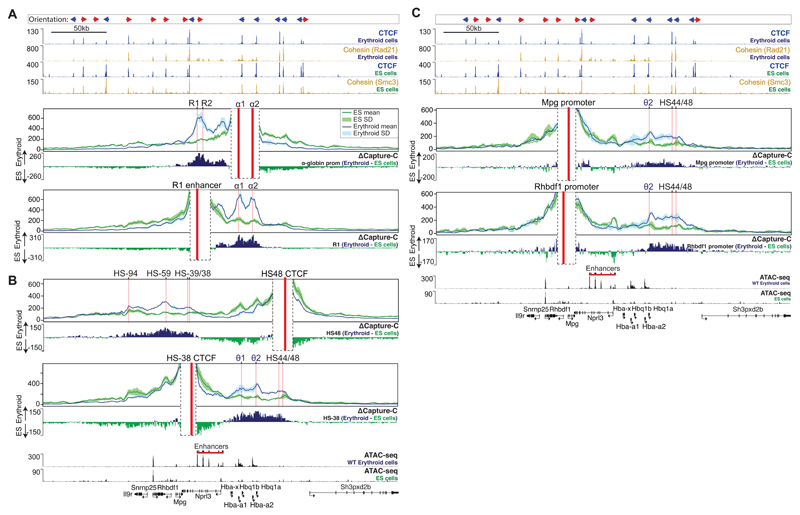
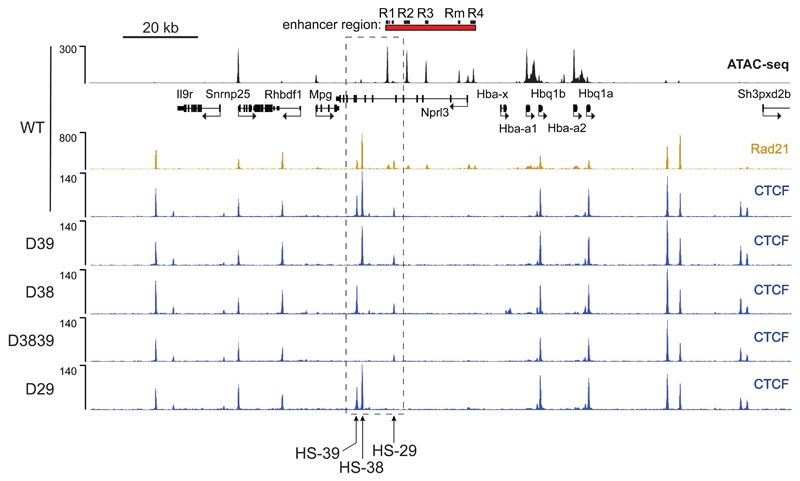
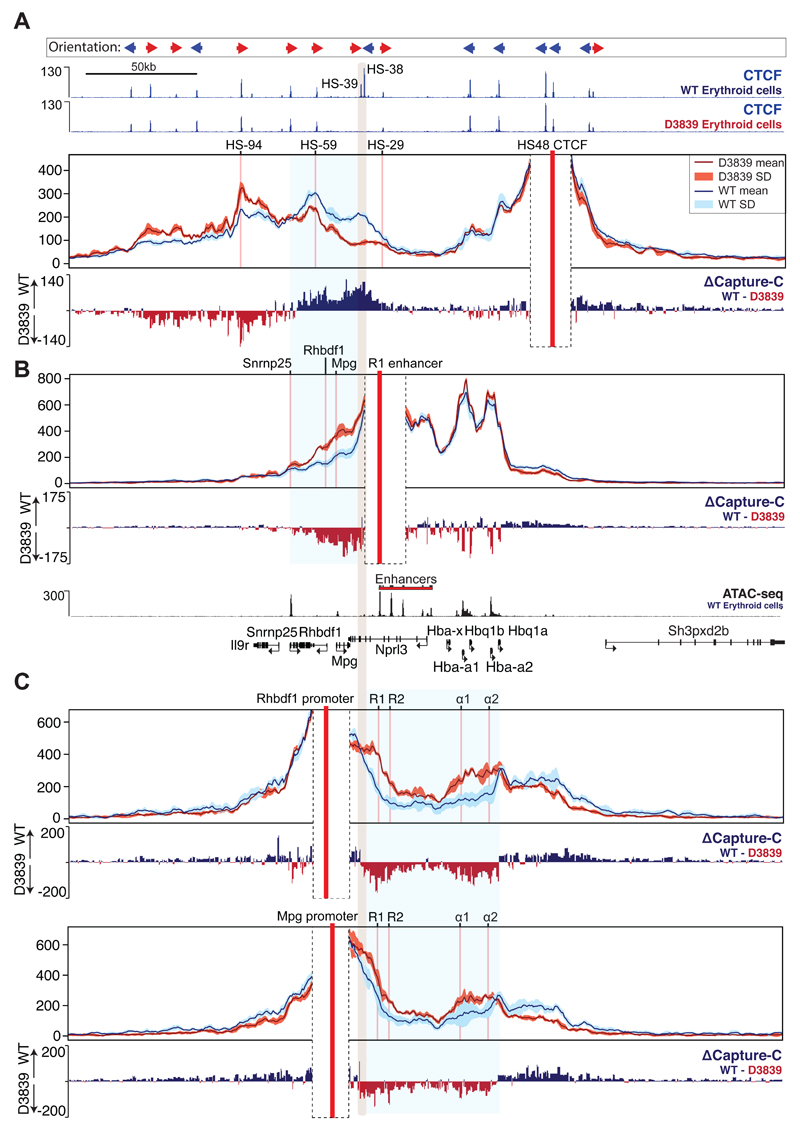
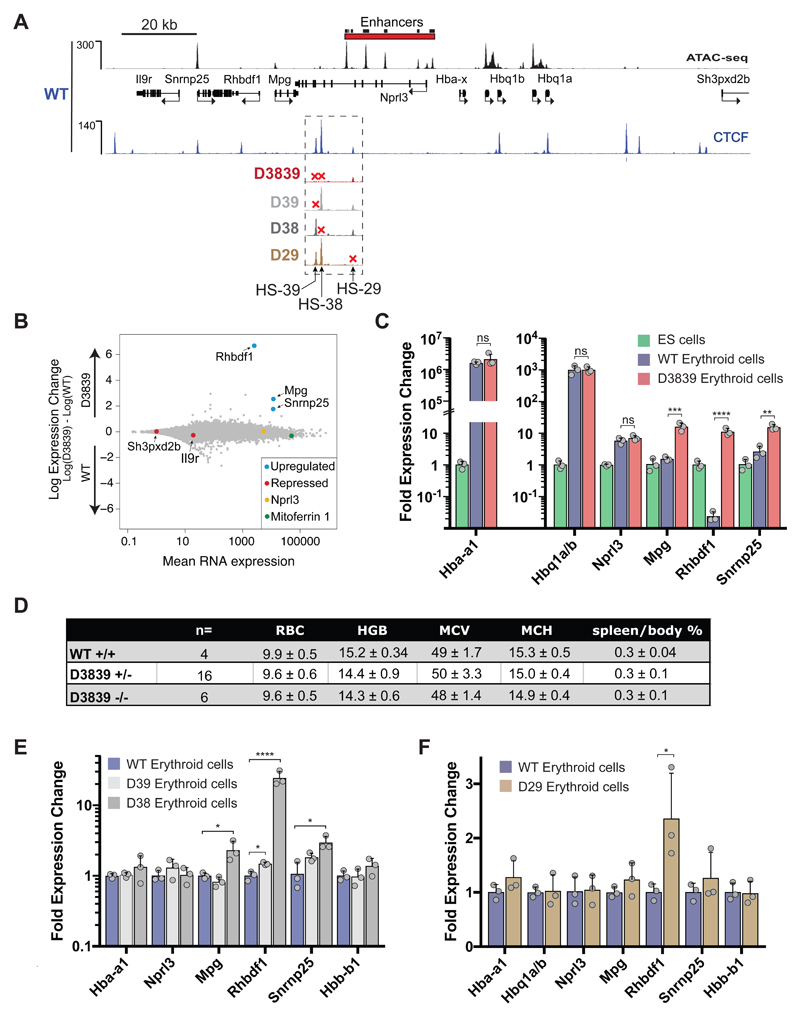
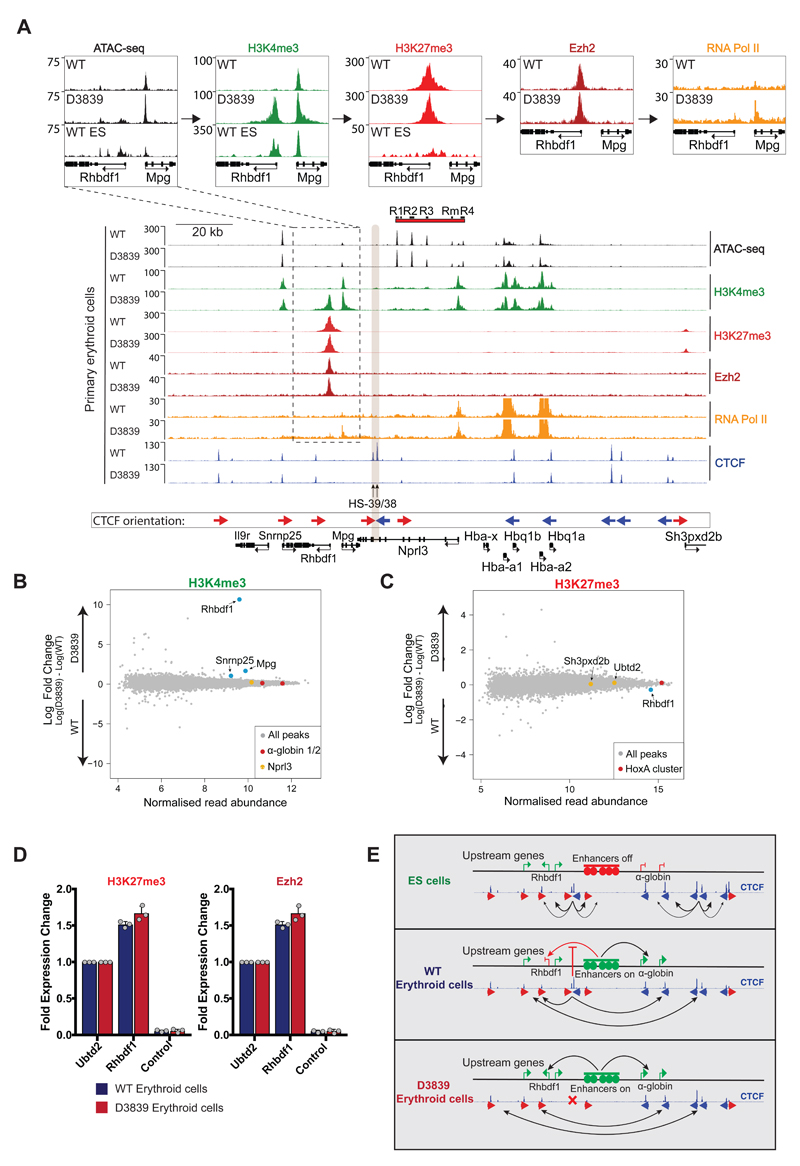
The genome is organized via CTCF-cohesin-binding sites, which partition chromosomes into 1-5 megabase (Mb) topologically associated domains (TADs), and further into smaller sub-domains (sub-TADs). Here we examined in vivo an ∼80 kb sub-TAD, containing the mouse α-globin gene cluster, lying within a ∼1 Mb TAD. We find that the sub-TAD is flanked by predominantly convergent CTCF-cohesin sites that are ubiquitously bound by CTCF but only interact during erythropoiesis, defining a self-interacting erythroid compartment. Whereas the α-globin regulatory elements normally act solely on promoters downstream of the enhancers, removal of a conserved upstream CTCF-cohesin boundary extends the sub-TAD to adjacent upstream CTCF-cohesin-binding sites. The α-globin enhancers now interact with the flanking chromatin, upregulating expression of genes within this extended sub-TAD. Rather than acting solely as a barrier to chromatin modification, CTCF-cohesin boundaries in this sub-TAD delimit the region of chromatin to which enhancers have access and within which they interact with receptive promoters.
Conflict of interest statement
The authors have no competing financial interests
Figures
Similar articles
-
Impact of 3D genome organization, guided by cohesin and CTCF looping, on sex-biased chromatin interactions and gene expression in mouse liver.Epigenetics Chromatin. 2020 Jul 17;13(1):30. doi: 10.1186/s13072-020-00350-y. Epigenetics Chromatin. 2020. PMID: 32680543 Free PMC article.
-
Activation of the alpha-globin gene expression correlates with dramatic upregulation of nearby non-globin genes and changes in local and large-scale chromatin spatial structure.Epigenetics Chromatin. 2017 Jul 11;10(1):35. doi: 10.1186/s13072-017-0142-4. Epigenetics Chromatin. 2017. PMID: 28693562 Free PMC article.
-
Computational prediction of CTCF/cohesin-based intra-TAD loops that insulate chromatin contacts and gene expression in mouse liver.Elife. 2018 May 14;7:e34077. doi: 10.7554/eLife.34077. Elife. 2018. PMID: 29757144 Free PMC article.
-
CTCF: the protein, the binding partners, the binding sites and their chromatin loops.Philos Trans R Soc Lond B Biol Sci. 2013 May 6;368(1620):20120369. doi: 10.1098/rstb.2012.0369. Print 2013. Philos Trans R Soc Lond B Biol Sci. 2013. PMID: 23650640 Free PMC article. Review.
-
Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation.J Biol Chem. 2012 Sep 7;287(37):30906-13. doi: 10.1074/jbc.R111.324962. Epub 2012 Sep 5. J Biol Chem. 2012. PMID: 22952237 Free PMC article. Review.
Cited by
-
LncRNAs are regulated by chromatin states and affect the skeletal muscle cell differentiation.Cell Prolif. 2020 Sep;53(9):e12879. doi: 10.1111/cpr.12879. Epub 2020 Aug 7. Cell Prolif. 2020. PMID: 32770602 Free PMC article.
-
Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements.Nat Commun. 2019 Sep 6;10(1):4063. doi: 10.1038/s41467-019-11955-7. Nat Commun. 2019. PMID: 31492858 Free PMC article.
-
The insulator functions of the Drosophila polydactyl C2H2 zinc finger protein CTCF: Necessity versus sufficiency.Sci Adv. 2020 Mar 25;6(13):eaaz3152. doi: 10.1126/sciadv.aaz3152. eCollection 2020 Mar. Sci Adv. 2020. PMID: 32232161 Free PMC article.
-
New insights emerge as antibody repertoire diversification meets chromosome conformation.F1000Res. 2019 Mar 28;8:F1000 Faculty Rev-347. doi: 10.12688/f1000research.17358.1. eCollection 2019. F1000Res. 2019. PMID: 30984378 Free PMC article. Review.
-
Folding Keratin Gene Clusters during Skin Regional Specification.Dev Cell. 2020 Jun 8;53(5):561-576.e9. doi: 10.1016/j.devcel.2020.05.007. Dev Cell. 2020. PMID: 32516596 Free PMC article.
References
-
- Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17:661–678. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
