

Enhancing the Oncolytic Activity of CD133-Targeted Measles Virus: Receptor Extension or Chimerism with Vesicular Stomatitis Virus Are Most Effective
- PMID: 28695108
- PMCID: PMC5483446
- DOI: 10.3389/fonc.2017.00127
Enhancing the Oncolytic Activity of CD133-Targeted Measles Virus: Receptor Extension or Chimerism with Vesicular Stomatitis Virus Are Most Effective
Abstract
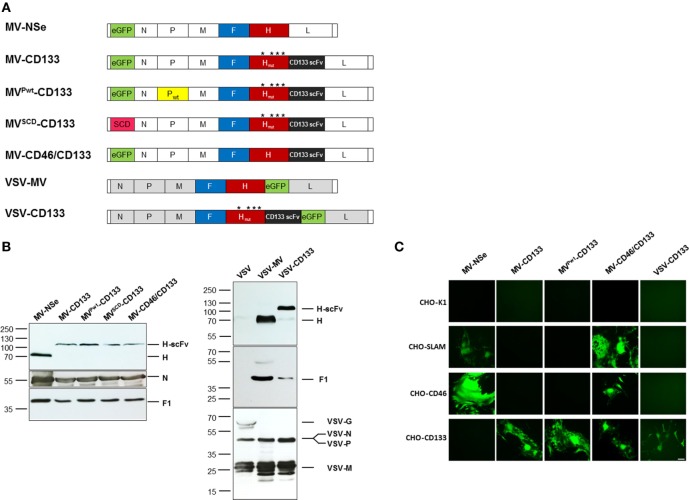
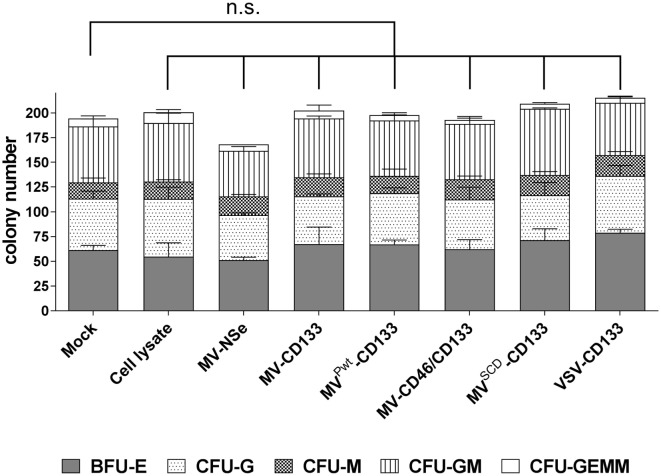
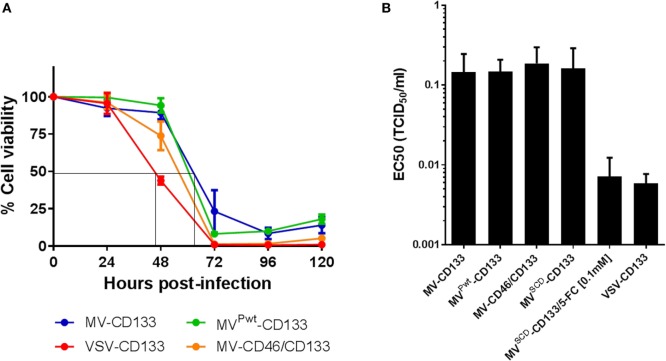
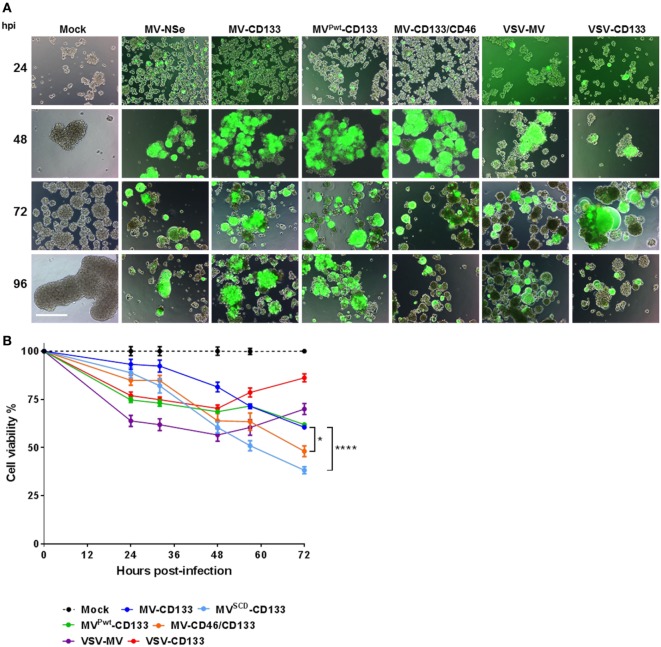
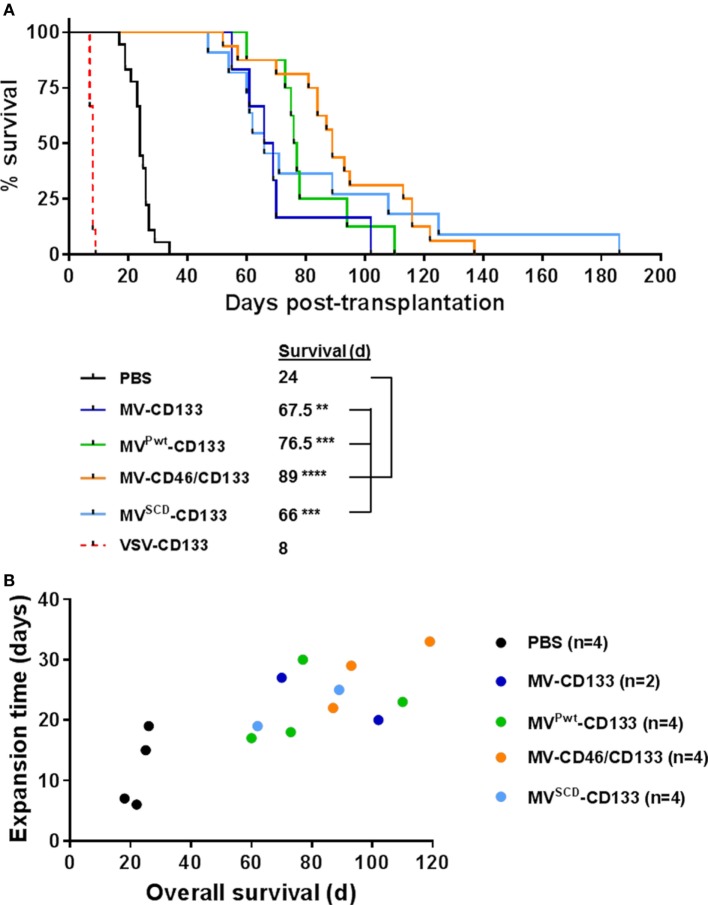
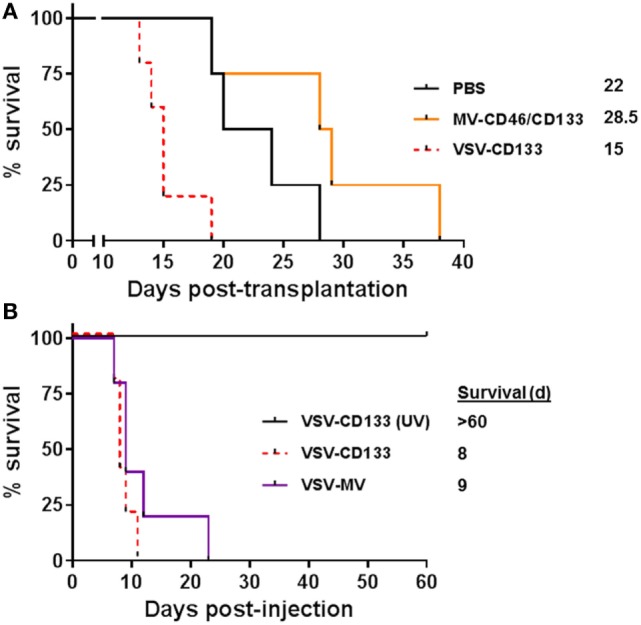
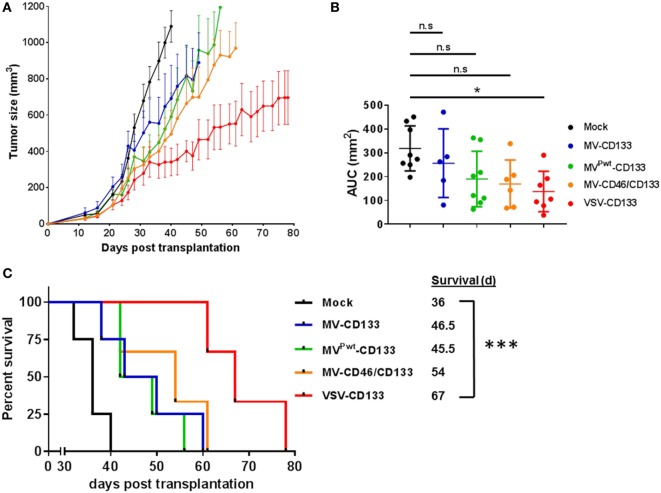
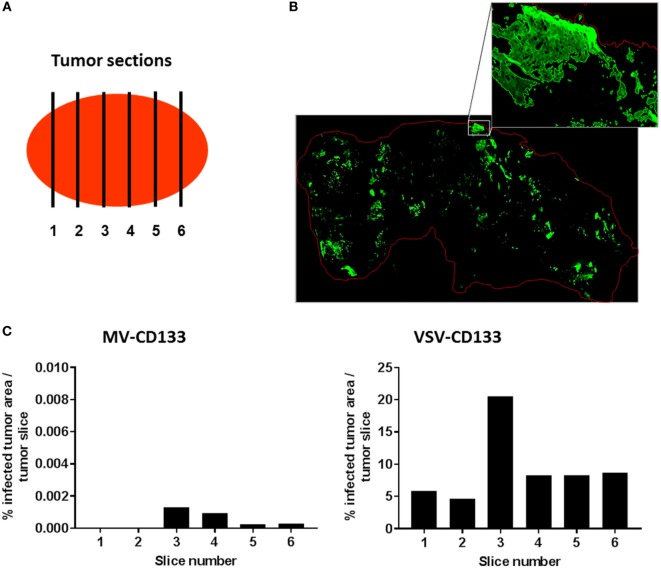
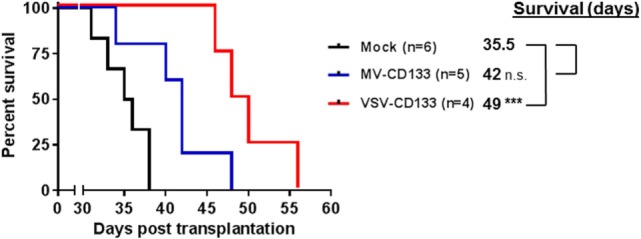
Therapy resistance and tumor recurrence are often linked to a small refractory and highly tumorigenic subpopulation of neoplastic cells, known as cancer stem cells (CSCs). A putative marker of CSCs is CD133 (prominin-1). We have previously described a CD133-targeted oncolytic measles virus (MV-CD133) as a promising approach to specifically eliminate CD133-positive tumor cells. Selectivity was introduced at the level of cell entry by an engineered MV hemagglutinin (H). The H protein was blinded for its native receptors and displayed a CD133-specific single-chain antibody fragment (scFv) as targeting domain. Interestingly, MV-CD133 was more active in killing CD133-positive tumors than the unmodified MV-NSe despite being highly selective for its target cells. To further enhance the antitumoral activity of MV-CD133, we here pursued arming technologies, receptor extension, and chimeras between MV-CD133 and vesicular stomatitis virus (VSV). All newly generated viruses including VSV-CD133 were highly selective in eliminating CD133-positive cells. MV-CD46/CD133 killed in addition CD133-negative cells being positive for the MV receptors. In an orthotopic glioma model, MV-CD46/CD133 and MVSCD-CD133, which encodes the super cytosine deaminase, were most effective. Notably, VSV-CD133 caused fatal neurotoxicity in this tumor model. Use of CD133 as receptor could be excluded as being causative. In a subcutaneous tumor model of hepatocellular cancer, VSV-CD133 revealed the most potent oncolytic activity and also significantly prolonged survival of the mice when injected intravenously. Compared to MV-CD133, VSV-CD133 infected a more than 104-fold larger area of the tumor within the same time period. Our data not only suggest new concepts and approaches toward enhancing the oncolytic activity of CD133-targeted oncolytic viruses but also raise awareness about careful toxicity testing of novel virus types.
Keywords: glioblastoma; hepatocellular carcinoma; prominin-1; tumorsphere; virotherapy.
Figures
Similar articles
-
Specific elimination of CD133+ tumor cells with targeted oncolytic measles virus.Cancer Res. 2013 Jan 15;73(2):865-74. doi: 10.1158/0008-5472.CAN-12-2221. Epub 2013 Jan 4. Cancer Res. 2013. PMID: 23293278
-
Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins.Hum Gene Ther. 2012 May;23(5):484-91. doi: 10.1089/hum.2011.146. Epub 2012 Feb 7. Hum Gene Ther. 2012. PMID: 22171635 Free PMC article.
-
CD30-targeted oncolytic viruses as novel therapeutic approach against classical Hodgkin lymphoma.Oncotarget. 2018 Jan 12;9(16):12971-12981. doi: 10.18632/oncotarget.24191. eCollection 2018 Feb 27. Oncotarget. 2018. PMID: 29560124 Free PMC article.
-
Measles to the Rescue: A Review of Oncolytic Measles Virus.Viruses. 2016 Oct 22;8(10):294. doi: 10.3390/v8100294. Viruses. 2016. PMID: 27782084 Free PMC article. Review.
-
VSV based virotherapy in ovarian cancer: the past, the present and …future?J Cancer. 2017 Jul 22;8(12):2369-2383. doi: 10.7150/jca.19473. eCollection 2017. J Cancer. 2017. PMID: 28819441 Free PMC article. Review.
Cited by
-
In vivo antitumor activity by dual stromal and tumor-targeted oncolytic measles viruses.Cancer Gene Ther. 2020 Dec;27(12):910-922. doi: 10.1038/s41417-020-0171-1. Epub 2020 Mar 31. Cancer Gene Ther. 2020. PMID: 32231231 Free PMC article.
-
Measles Virus-Based Treatments Trigger a Pro-inflammatory Cascade and a Distinctive Immunopeptidome in Glioblastoma.Mol Ther Oncolytics. 2018 Dec 31;12:147-161. doi: 10.1016/j.omto.2018.12.010. eCollection 2019 Mar 29. Mol Ther Oncolytics. 2018. PMID: 30775418 Free PMC article.
-
DCLK1 Expression in Colorectal Polyps Increases with the Severity of Dysplasia.In Vivo. 2018 Mar-Apr;32(2):365-371. doi: 10.21873/invivo.11247. In Vivo. 2018. PMID: 29475922 Free PMC article.
-
Resistance Mechanisms Influencing Oncolytic Virotherapy, a Systematic Analysis.Vaccines (Basel). 2021 Oct 12;9(10):1166. doi: 10.3390/vaccines9101166. Vaccines (Basel). 2021. PMID: 34696274 Free PMC article. Review.
-
New frontiers in oncolytic viruses: optimizing and selecting for virus strains with improved efficacy.Biologics. 2018 Feb 9;12:43-60. doi: 10.2147/BTT.S140114. eCollection 2018. Biologics. 2018. PMID: 29445265 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials