

ML290 is a biased allosteric agonist at the relaxin receptor RXFP1
- PMID: 28592882
- PMCID: PMC5462828
- DOI: 10.1038/s41598-017-02916-5
ML290 is a biased allosteric agonist at the relaxin receptor RXFP1
Abstract
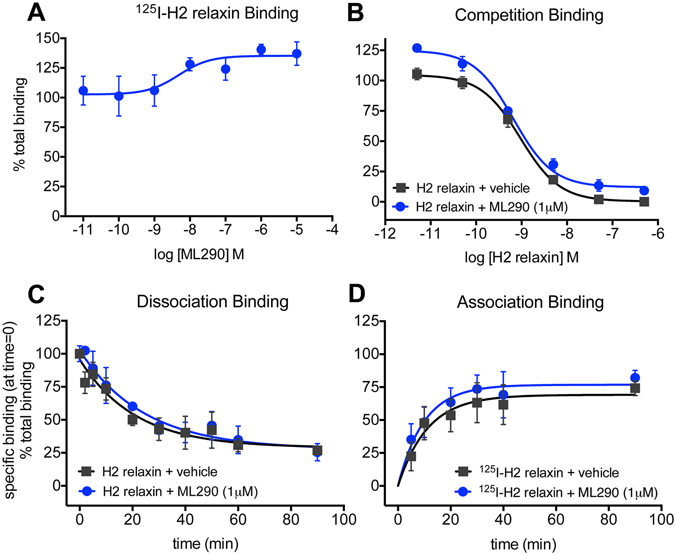
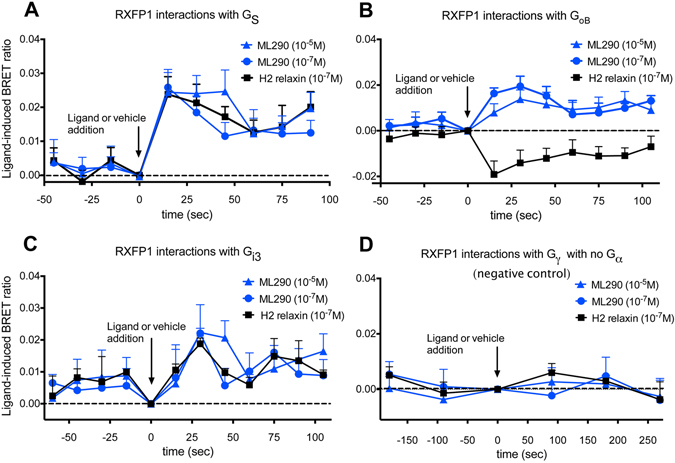
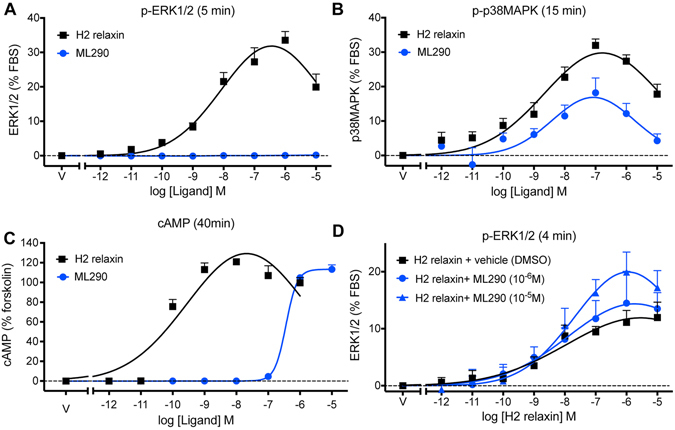
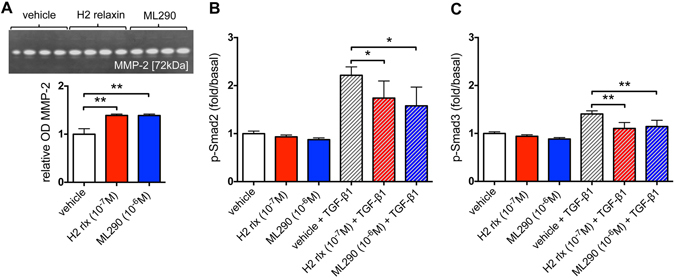
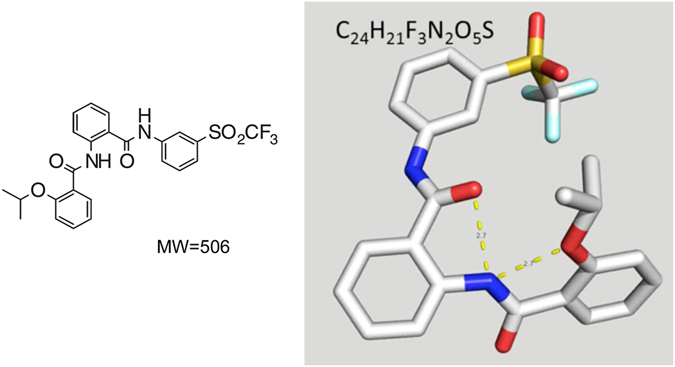
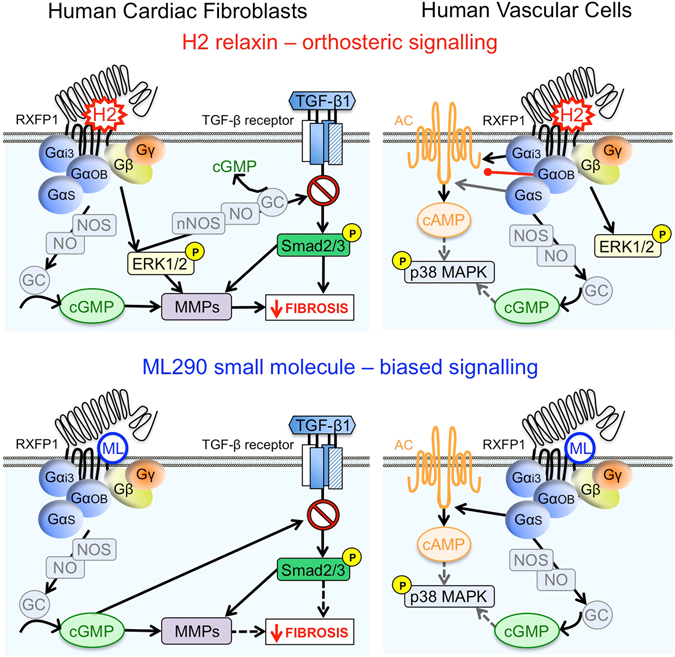
Activation of the relaxin receptor RXFP1 has been associated with improved survival in acute heart failure. ML290 is a small molecule RXFP1 agonist with simple structure, long half-life and high stability. Here we demonstrate that ML290 is a biased agonist in human cells expressing RXFP1 with long-term beneficial actions on markers of fibrosis in human cardiac fibroblasts (HCFs). ML290 did not directly compete with orthosteric relaxin binding and did not affect binding kinetics, but did increase binding to RXFP1. In HEK-RXFP1 cells, ML290 stimulated cAMP accumulation and p38MAPK phosphorylation but not cGMP accumulation or ERK1/2 phosphorylation although prior addition of ML290 increased p-ERK1/2 responses to relaxin. In human primary vascular endothelial and smooth muscle cells that endogenously express RXFP1, ML290 increased both cAMP and cGMP accumulation but not p-ERK1/2. In HCFs, ML290 increased cGMP accumulation but did not affect p-ERK1/2 and given chronically activated MMP-2 expression and inhibited TGF-β1-induced Smad2 and Smad3 phosphorylation. In vascular cells, ML290 was 10x more potent for cGMP accumulation and p-p38MAPK than for cAMP accumulation. ML290 caused strong coupling of RXFP1 to Gαs and GαoB but weak coupling to Gαi3. ML290 exhibited signalling bias at RXFP1 possessing a signalling profile indicative of vasodilator and anti-fibrotic properties.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
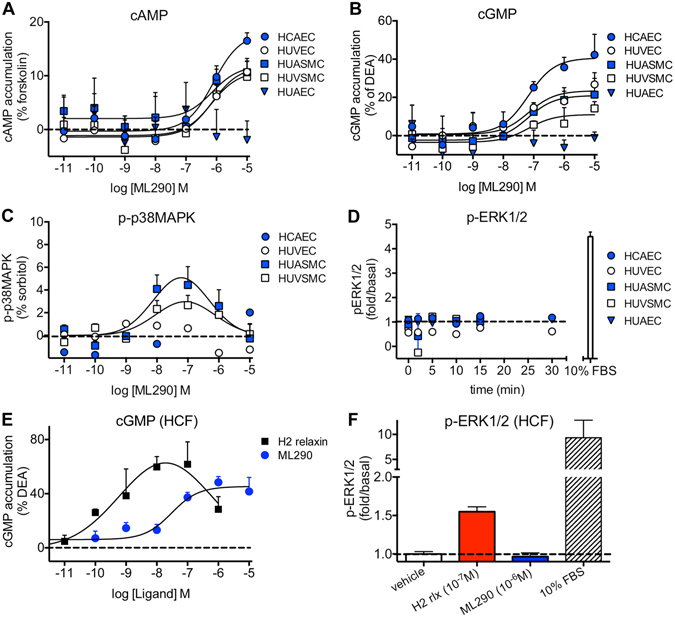
 , n = 7), HUVECs (
, n = 7), HUVECs ( , n = 7), HUASMCs (
, n = 7), HUASMCs ( , n = 5), HUVSMCs (
, n = 5), HUVSMCs ( , n = 4) but not in HUAECs that do not express cell surface RXFP1 (
, n = 4) but not in HUAECs that do not express cell surface RXFP1 ( , n = 4); in (B) ML290 (30 min) also increased cGMP accumulation in HCAECs (n = 7), HUVECs (n = 7), HUASMCs (n = 7), HUVSMCs (n = 4) but not in HUAECs (n = 4); In (C) ML290 increased p-p38MAPK (15 min) but only in HUASMC and HUVSMCs (n = 3); In (D) ML290 did not cause ERK1/2 phosphorylation in HCAECs (n = 3), HUVECs (n = 3), HUASMCs (n = 3), HUVSMCs (n = 3) or HUAECs (n = 3). In HCFs, H2 relaxin activated cGMP accumulation in a concentration-dependent manner ((E) n = 5) (40 min) and p-ERK1/2 ((F) n = 3) (5 min). ML290 did not activate p-ERK1/2 (F; n = 3) but did induce cGMP accumulation (E; n = 5) in a concentration-dependent manner albeit with lower potency than H2 relaxin. Data shown are mean ± SEM of ‘n’ independent experiments.
, n = 4); in (B) ML290 (30 min) also increased cGMP accumulation in HCAECs (n = 7), HUVECs (n = 7), HUASMCs (n = 7), HUVSMCs (n = 4) but not in HUAECs (n = 4); In (C) ML290 increased p-p38MAPK (15 min) but only in HUASMC and HUVSMCs (n = 3); In (D) ML290 did not cause ERK1/2 phosphorylation in HCAECs (n = 3), HUVECs (n = 3), HUASMCs (n = 3), HUVSMCs (n = 3) or HUAECs (n = 3). In HCFs, H2 relaxin activated cGMP accumulation in a concentration-dependent manner ((E) n = 5) (40 min) and p-ERK1/2 ((F) n = 3) (5 min). ML290 did not activate p-ERK1/2 (F; n = 3) but did induce cGMP accumulation (E; n = 5) in a concentration-dependent manner albeit with lower potency than H2 relaxin. Data shown are mean ± SEM of ‘n’ independent experiments.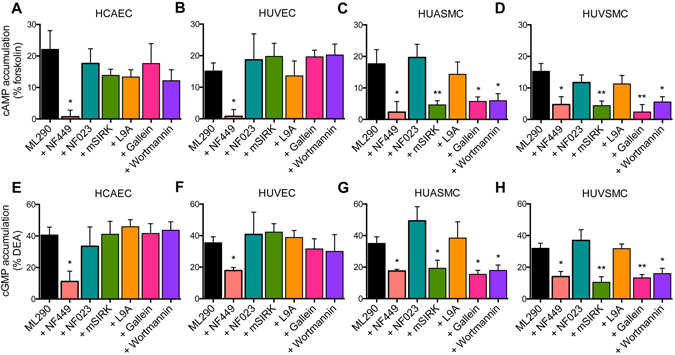
Similar articles
-
Structural Insights into the Activation of Human Relaxin Family Peptide Receptor 1 by Small-Molecule Agonists.Biochemistry. 2016 Mar 29;55(12):1772-83. doi: 10.1021/acs.biochem.5b01195. Epub 2016 Mar 4. Biochemistry. 2016. PMID: 26866459 Free PMC article.
-
Activation of Relaxin Family Receptor 1 from Different Mammalian Species by Relaxin Peptide and Small-Molecule Agonist ML290.Front Endocrinol (Lausanne). 2015 Aug 17;6:128. doi: 10.3389/fendo.2015.00128. eCollection 2015. Front Endocrinol (Lausanne). 2015. PMID: 26347712 Free PMC article.
-
Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP signaling by distinct mechanisms.Mol Pharmacol. 2006 Jul;70(1):214-26. doi: 10.1124/mol.105.021691. Epub 2006 Mar 28. Mol Pharmacol. 2006. PMID: 16569707
-
International Union of Basic and Clinical Pharmacology. XCV. Recent advances in the understanding of the pharmacology and biological roles of relaxin family peptide receptors 1-4, the receptors for relaxin family peptides.Pharmacol Rev. 2015;67(2):389-440. doi: 10.1124/pr.114.009472. Pharmacol Rev. 2015. PMID: 25761609 Free PMC article. Review.
-
Relaxin family peptide receptors--former orphans reunite with their parent ligands to activate multiple signalling pathways.Br J Pharmacol. 2007 Mar;150(6):677-91. doi: 10.1038/sj.bjp.0707140. Epub 2007 Feb 12. Br J Pharmacol. 2007. PMID: 17293890 Free PMC article. Review.
Cited by
-
Discovery of small molecule agonists of the Relaxin Family Peptide Receptor 2.Commun Biol. 2022 Nov 4;5(1):1183. doi: 10.1038/s42003-022-04143-9. Commun Biol. 2022. PMID: 36333465 Free PMC article.
-
Relaxin and fibrosis: Emerging targets, challenges, and future directions.Mol Cell Endocrinol. 2019 May 1;487:66-74. doi: 10.1016/j.mce.2019.02.005. Epub 2019 Feb 14. Mol Cell Endocrinol. 2019. PMID: 30772373 Free PMC article. Review.
-
Optimization of the first small-molecule relaxin/insulin-like family peptide receptor (RXFP1) agonists: Activation results in an antifibrotic gene expression profile.Eur J Med Chem. 2018 Aug 5;156:79-92. doi: 10.1016/j.ejmech.2018.06.008. Epub 2018 Jun 7. Eur J Med Chem. 2018. PMID: 30006176 Free PMC article.
-
Relaxin as a treatment for musculoskeletal fibrosis: What we know and future directions.Biochem Pharmacol. 2024 Jul;225:116273. doi: 10.1016/j.bcp.2024.116273. Epub 2024 May 8. Biochem Pharmacol. 2024. PMID: 38729446 Review.
-
Characterization of a new potent and long-lasting single chain peptide agonist of RXFP1 in cells and in vivo translational models.Sci Rep. 2022 Nov 28;12(1):20435. doi: 10.1038/s41598-022-24716-2. Sci Rep. 2022. PMID: 36443381 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
