

Zika virus evolution and spread in the Americas
- PMID: 28538734
- PMCID: PMC5563848
- DOI: 10.1038/nature22402
Zika virus evolution and spread in the Americas
Abstract
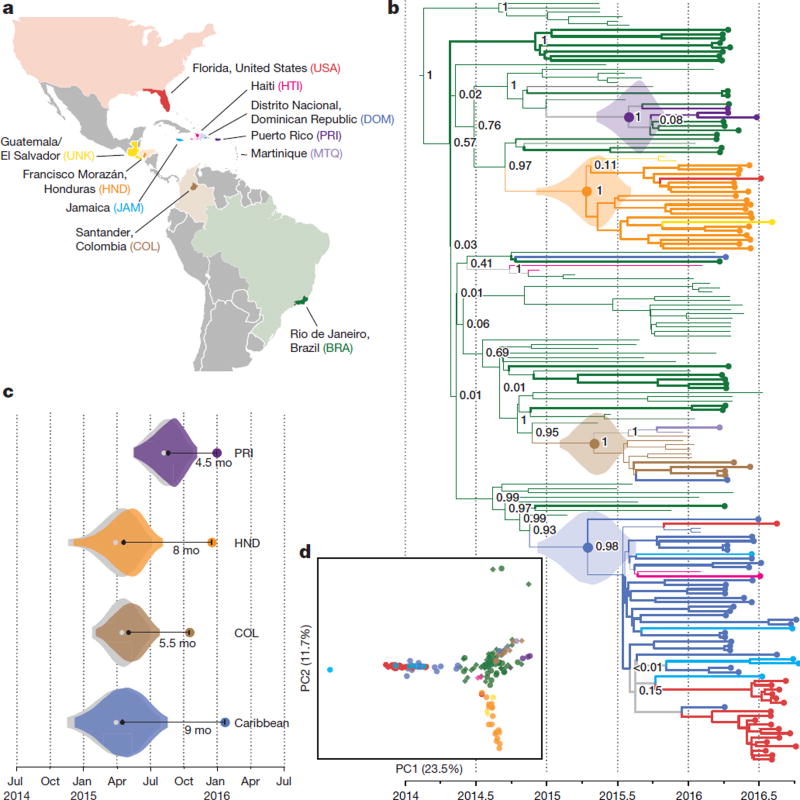
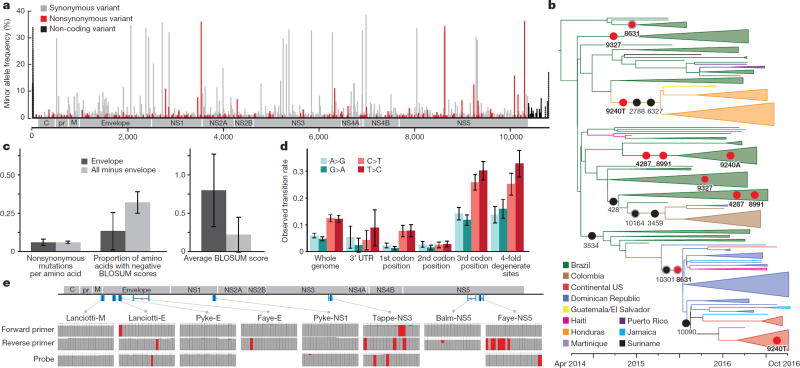
Although the recent Zika virus (ZIKV) epidemic in the Americas and its link to birth defects have attracted a great deal of attention, much remains unknown about ZIKV disease epidemiology and ZIKV evolution, in part owing to a lack of genomic data. Here we address this gap in knowledge by using multiple sequencing approaches to generate 110 ZIKV genomes from clinical and mosquito samples from 10 countries and territories, greatly expanding the observed viral genetic diversity from this outbreak. We analysed the timing and patterns of introductions into distinct geographic regions; our phylogenetic evidence suggests rapid expansion of the outbreak in Brazil and multiple introductions of outbreak strains into Puerto Rico, Honduras, Colombia, other Caribbean islands, and the continental United States. We find that ZIKV circulated undetected in multiple regions for many months before the first locally transmitted cases were confirmed, highlighting the importance of surveillance of viral infections. We identify mutations with possible functional implications for ZIKV biology and pathogenesis, as well as those that might be relevant to the effectiveness of diagnostic tests.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Epidemiology: Molecular mapping of Zika spread.Nature. 2017 Jun 15;546(7658):355-357. doi: 10.1038/nature22495. Epub 2017 May 24. Nature. 2017. PMID: 28538722 No abstract available.
Similar articles
-
Genomic epidemiology supports multiple introductions and cryptic transmission of Zika virus in Colombia.BMC Infect Dis. 2019 Nov 12;19(1):963. doi: 10.1186/s12879-019-4566-2. BMC Infect Dis. 2019. PMID: 31718580 Free PMC article.
-
Genomic epidemiology reveals multiple introductions of Zika virus into the United States.Nature. 2017 Jun 15;546(7658):401-405. doi: 10.1038/nature22400. Epub 2017 May 24. Nature. 2017. PMID: 28538723 Free PMC article.
-
Zika virus in the Americas: Early epidemiological and genetic findings.Science. 2016 Apr 15;352(6283):345-349. doi: 10.1126/science.aaf5036. Epub 2016 Mar 24. Science. 2016. PMID: 27013429 Free PMC article.
-
Zika virus-spread, epidemiology, genome, transmission cycle, clinical manifestation, associated challenges, vaccine and antiviral drug development.Virology. 2020 Apr;543:34-42. doi: 10.1016/j.virol.2020.01.015. Epub 2020 Feb 2. Virology. 2020. PMID: 32056845 Review.
-
The Asian Lineage of Zika Virus: Transmission and Evolution in Asia and the Americas.Virol Sin. 2019 Feb;34(1):1-8. doi: 10.1007/s12250-018-0078-2. Epub 2019 Jan 25. Virol Sin. 2019. PMID: 30684211 Free PMC article. Review.
Cited by
-
Comparative mutational analysis of the Zika virus genome from different geographical locations and its effect on the efficacy of Zika virus-specific neutralizing antibodies.Front Microbiol. 2023 Feb 22;14:1098323. doi: 10.3389/fmicb.2023.1098323. eCollection 2023. Front Microbiol. 2023. PMID: 36910181 Free PMC article.
-
Impression Cytology Is a Non-invasive and Effective Method for Ocular Cell Retrieval of Zika Infected Babies: Perspectives in OMIC Studies.Front Mol Neurosci. 2019 Dec 5;12:279. doi: 10.3389/fnmol.2019.00279. eCollection 2019. Front Mol Neurosci. 2019. PMID: 31866819 Free PMC article.
-
A reverse-transcription/RNase H based protocol for depletion of mosquito ribosomal RNA facilitates viral intrahost evolution analysis, transcriptomics and pathogen discovery.Virology. 2019 Feb;528:181-197. doi: 10.1016/j.virol.2018.12.020. Epub 2018 Dec 30. Virology. 2019. PMID: 30616207 Free PMC article.
-
An Evolutionary Insight into Zika Virus Strains Isolated in the Latin American Region.Viruses. 2018 Dec 8;10(12):698. doi: 10.3390/v10120698. Viruses. 2018. PMID: 30544785 Free PMC article.
-
Spatial diffusion of the 2015-2016 Zika, dengue and chikungunya epidemics in Rio de Janeiro Municipality, Brazil.Epidemiol Infect. 2019 Jan;147:e237. doi: 10.1017/S0950268819001250. Epidemiol Infect. 2019. PMID: 31364556 Free PMC article.
References
-
- World Health Organization. Zika situation report: Zika virus, Microcephaly and Guillain–Barré syndrome. 2017 http://who.int/emergencies/zika-virus/situation-report/2-february-2017/en/
-
- de Vigilância em Saúde S. Protocolo de Vigilância e Resposta à Ocorrência de Microcefalia. Ministério da Saúde Brasília; 2016.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
