

NF-κB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases
- PMID: 28469620
- PMCID: PMC5395695
- DOI: 10.3389/fimmu.2017.00399
NF-κB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases
Abstract
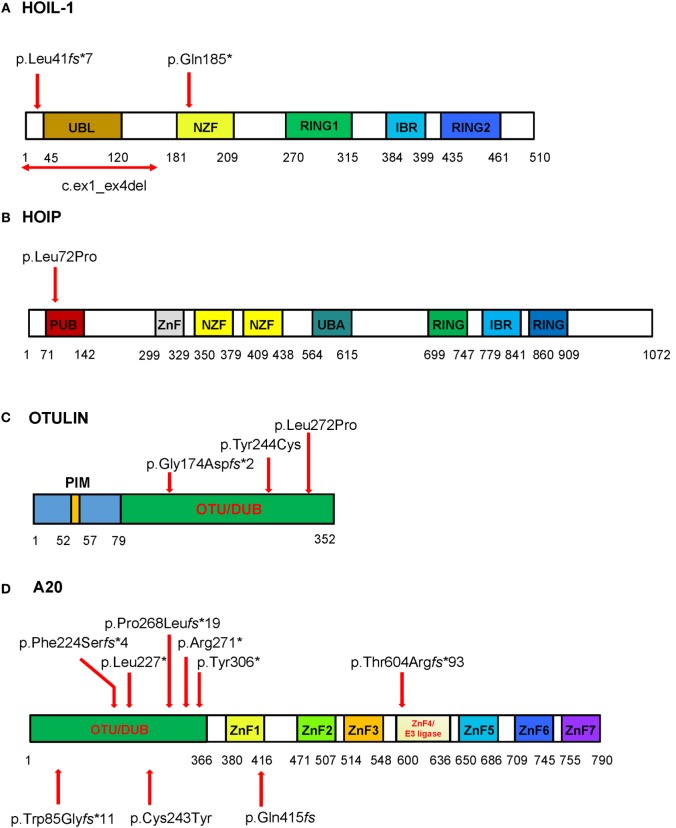
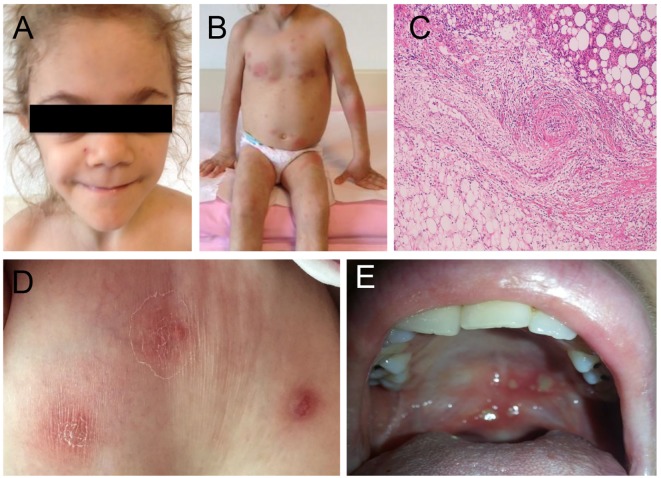
Autoinflammatory diseases are caused by defects in genes that regulate the innate immunity. Recently, the scope of autoinflammation has been broadened to include diseases that result from dysregulations in protein modifications by the highly conserved ubiquitin (Ub) peptides. Thus far these diseases consist of linear ubiquitin chain assembly complex (LUBAC) and OTULIN deficiencies, and haploinsufficiency of A20. The LUBAC is critical for linear ubiquitination of key signaling molecules in immune response pathways, while deubiquitinase enzymes, OTULIN and TNFAIP3/A20, reverse the effects of ubiquitination by hydrolyzing linear (Met1) and Lys63 (K63) Ub moieties, respectively, from conjugated proteins. Consequently, OTULIN or A20-deficient cells have an excess of Met1 or K63 Ub chains on NEMO, RIPK1, and other target substrates, which lead to constitutive activation of the NF-kB pathway. Mutant cells produce elevated levels of many proinflammatory cytokines and respond to therapy with cytokine inhibitors. Patients with an impairment in LUBAC stability have compromised NF-kB responses in non-immune cells such as fibroblasts, while their monocytes are hyperresponsive to IL-1β. Discoveries of germline mutations in enzymes that regulate protein modifications by Ub define a new category of autoinflammatory diseases caused by upregulations in the NF-kB signaling. The primary aim of this review is to summarize the latest developments in our understanding of the etiology of autoinflammation.
Keywords: LUBAC deficiency; OTULIN; TNFAIP3/A20; haploinsufficiency of A20; linear ubiquitin chain assembly complex; otulipenia/otulin-related autoinflammatory syndrome.
Figures

Similar articles
-
An Update on Autoinflammatory Diseases: Relopathies.Curr Rheumatol Rep. 2018 May 30;20(7):39. doi: 10.1007/s11926-018-0749-x. Curr Rheumatol Rep. 2018. PMID: 29846841 Review.
-
CYLD Limits Lys63- and Met1-Linked Ubiquitin at Receptor Complexes to Regulate Innate Immune Signaling.Cell Rep. 2016 Mar 29;14(12):2846-58. doi: 10.1016/j.celrep.2016.02.062. Epub 2016 Mar 17. Cell Rep. 2016. PMID: 26997266 Free PMC article.
-
OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin.Cell. 2013 Jun 6;153(6):1312-26. doi: 10.1016/j.cell.2013.05.014. Cell. 2013. PMID: 23746843 Free PMC article.
-
Linear ubiquitination-mediated NF-κB regulation and its related disorders.J Biochem. 2013 Oct;154(4):313-23. doi: 10.1093/jb/mvt079. Epub 2013 Aug 21. J Biochem. 2013. PMID: 23969028 Review.
-
OTULIN restricts Met1-linked ubiquitination to control innate immune signaling.Mol Cell. 2013 Jun 27;50(6):818-830. doi: 10.1016/j.molcel.2013.06.004. Mol Cell. 2013. PMID: 23806334 Free PMC article.
Cited by
-
A20 haploinsufficiency in a neonate caused by a large deletion on chromosome 6q.Pediatr Rheumatol Online J. 2024 Jan 5;22(1):12. doi: 10.1186/s12969-023-00947-z. Pediatr Rheumatol Online J. 2024. PMID: 38183052 Free PMC article. Review.
-
Hereditary systemic autoinflammatory diseases and Schnitzler's syndrome.Rheumatology (Oxford). 2019 Nov 1;58(Suppl 6):vi31-vi43. doi: 10.1093/rheumatology/kez448. Rheumatology (Oxford). 2019. PMID: 31769858 Free PMC article. Review.
-
Biochemistry of Autoinflammatory Diseases: Catalyzing Monogenic Disease.Front Immunol. 2019 Jan 31;10:101. doi: 10.3389/fimmu.2019.00101. eCollection 2019. Front Immunol. 2019. PMID: 30766537 Free PMC article. Review.
-
The genetics of macrophage activation syndrome.Genes Immun. 2020 May;21(3):169-181. doi: 10.1038/s41435-020-0098-4. Epub 2020 Apr 15. Genes Immun. 2020. PMID: 32291394 Review.
-
Vasculitis associated with VEXAS syndrome: A literature review.Front Med (Lausanne). 2022 Aug 15;9:983939. doi: 10.3389/fmed.2022.983939. eCollection 2022. Front Med (Lausanne). 2022. PMID: 36045928 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous