

Contrasting evolutionary genome dynamics between domesticated and wild yeasts
- PMID: 28416820
- PMCID: PMC5446901
- DOI: 10.1038/ng.3847
Contrasting evolutionary genome dynamics between domesticated and wild yeasts
Abstract
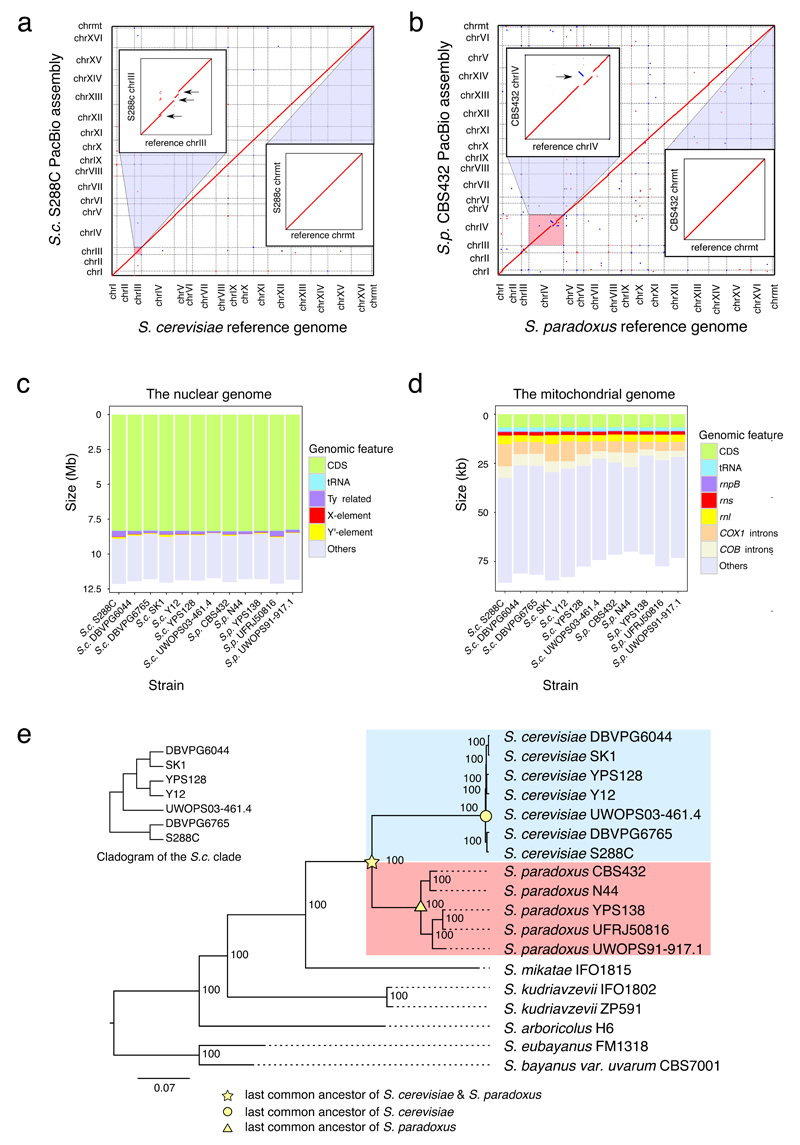
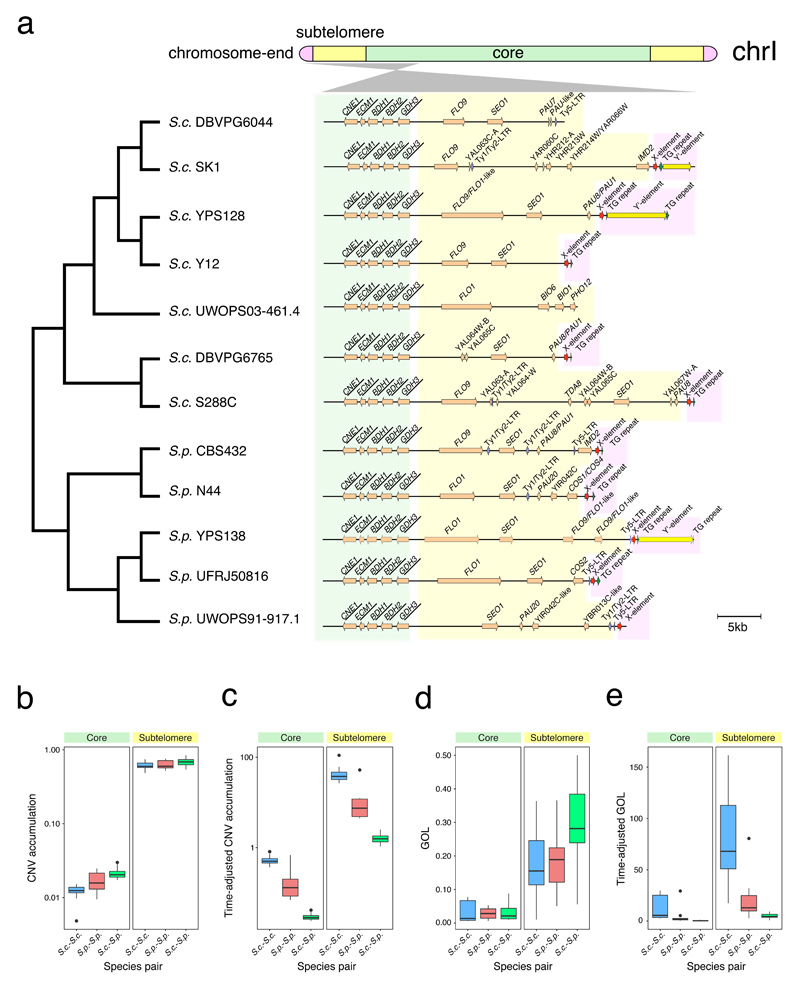
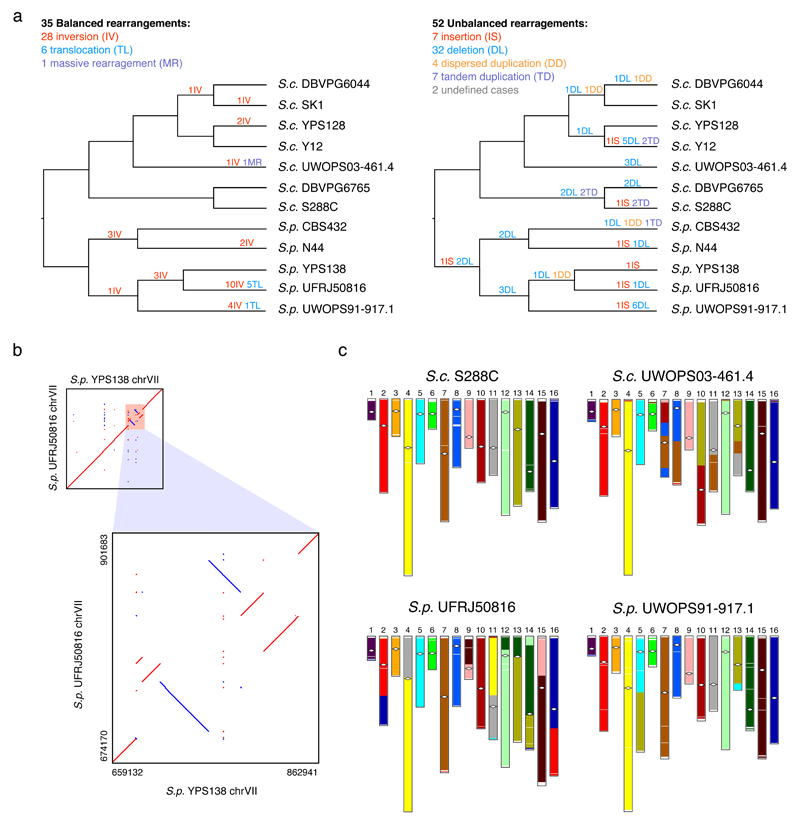
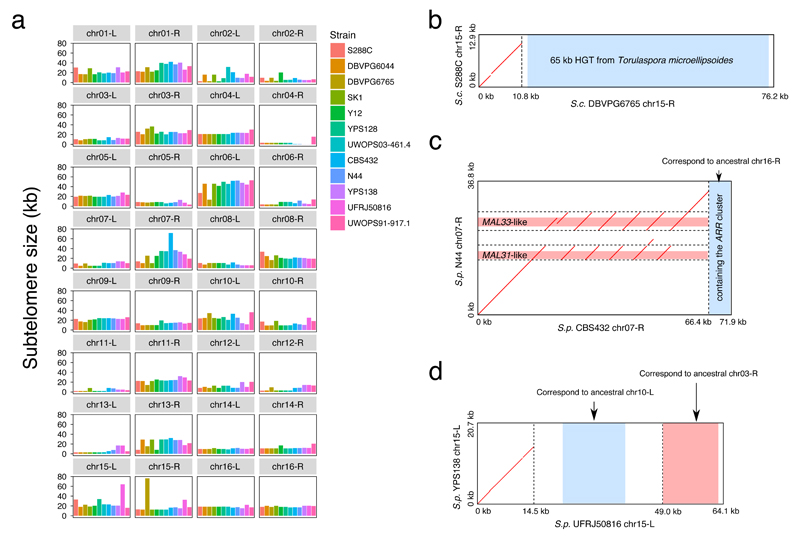
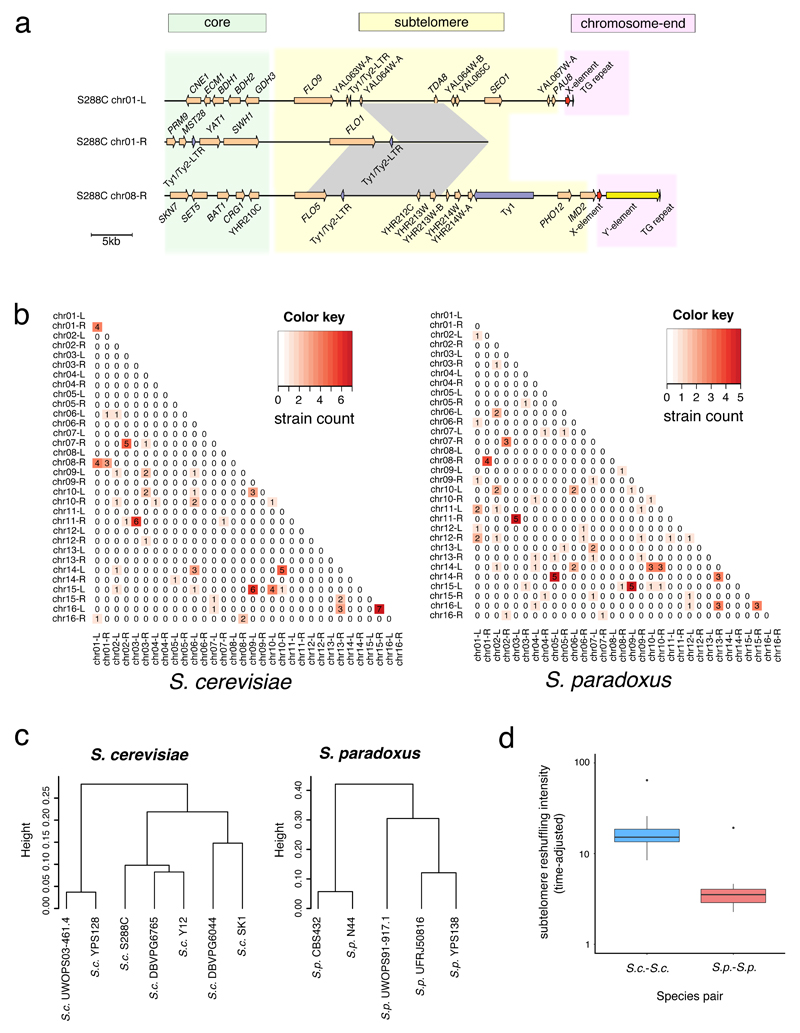
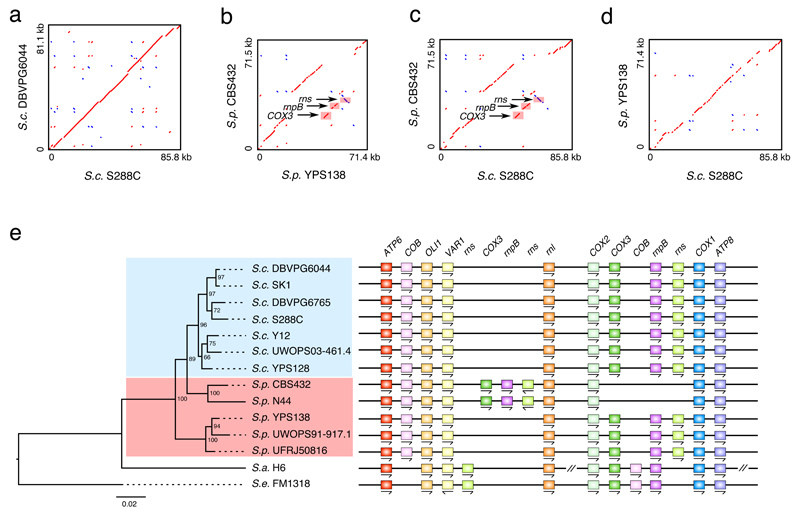
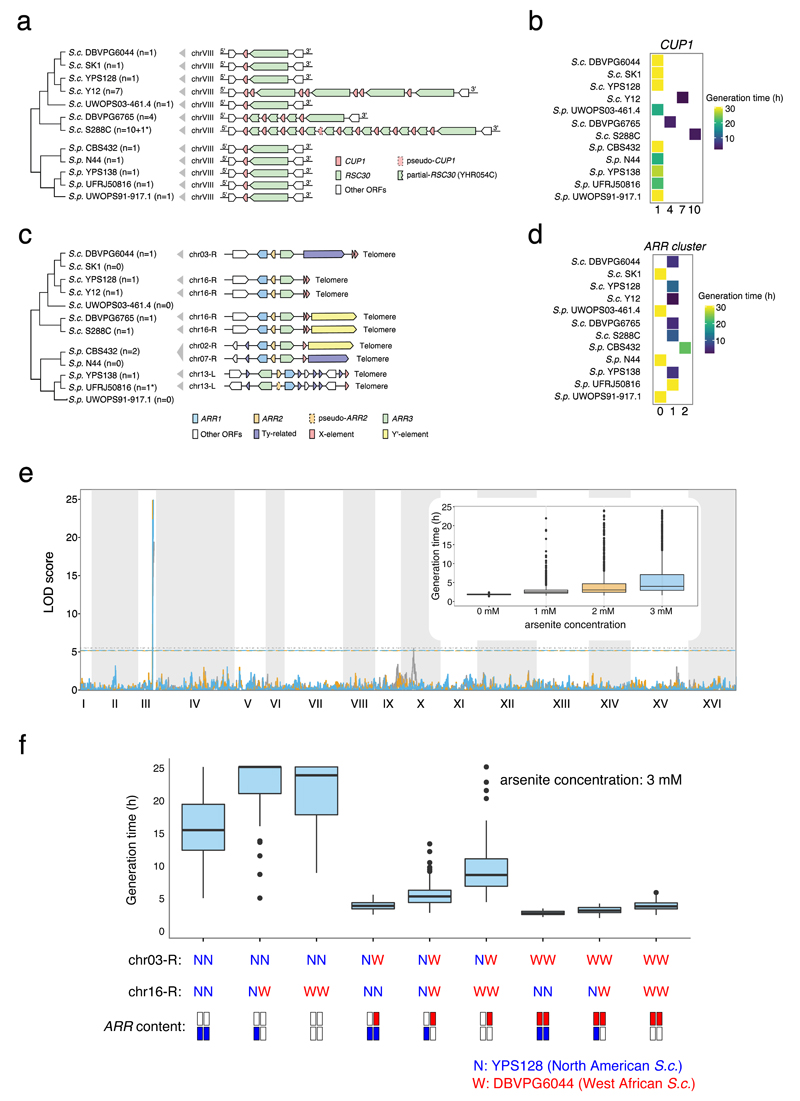
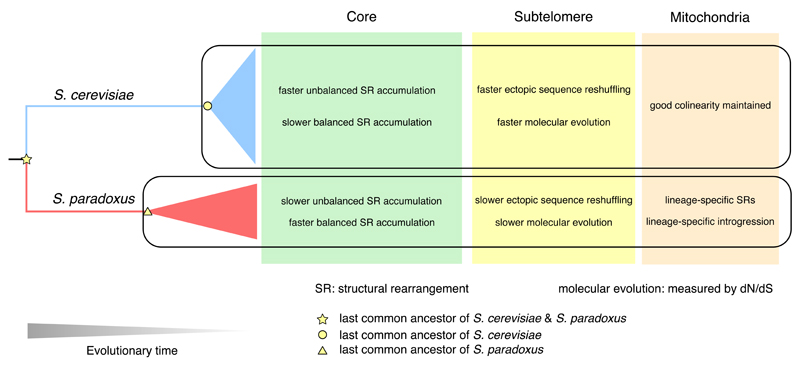
Structural rearrangements have long been recognized as an important source of genetic variation, with implications in phenotypic diversity and disease, yet their detailed evolutionary dynamics remain elusive. Here we use long-read sequencing to generate end-to-end genome assemblies for 12 strains representing major subpopulations of the partially domesticated yeast Saccharomyces cerevisiae and its wild relative Saccharomyces paradoxus. These population-level high-quality genomes with comprehensive annotation enable precise definition of chromosomal boundaries between cores and subtelomeres and a high-resolution view of evolutionary genome dynamics. In chromosomal cores, S. paradoxus shows faster accumulation of balanced rearrangements (inversions, reciprocal translocations and transpositions), whereas S. cerevisiae accumulates unbalanced rearrangements (novel insertions, deletions and duplications) more rapidly. In subtelomeres, both species show extensive interchromosomal reshuffling, with a higher tempo in S. cerevisiae. Such striking contrasts between wild and domesticated yeasts are likely to reflect the influence of human activities on structural genome evolution.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures
Similar articles
-
Evolution of gene order in the genomes of two related yeast species.Genome Res. 2001 Dec;11(12):2009-19. doi: 10.1101/gr.212701. Genome Res. 2001. PMID: 11731490
-
The Genome Sequence of Saccharomyces eubayanus and the Domestication of Lager-Brewing Yeasts.Mol Biol Evol. 2015 Nov;32(11):2818-31. doi: 10.1093/molbev/msv168. Epub 2015 Aug 11. Mol Biol Evol. 2015. PMID: 26269586 Free PMC article.
-
The genomes of fermentative Saccharomyces.C R Biol. 2011 Aug-Sep;334(8-9):687-93. doi: 10.1016/j.crvi.2011.05.019. Epub 2011 Jul 1. C R Biol. 2011. PMID: 21819951 Review.
-
Population genomics of domestic and wild yeasts.Nature. 2009 Mar 19;458(7236):337-41. doi: 10.1038/nature07743. Epub 2009 Feb 11. Nature. 2009. PMID: 19212322 Free PMC article.
-
Yeast evolution and comparative genomics.Annu Rev Microbiol. 2005;59:135-53. doi: 10.1146/annurev.micro.59.030804.121400. Annu Rev Microbiol. 2005. PMID: 15877535 Review.
Cited by
-
Revisiting the Taxonomic Synonyms and Populations of Saccharomyces cerevisiae-Phylogeny, Phenotypes, Ecology and Domestication.Microorganisms. 2020 Jun 15;8(6):903. doi: 10.3390/microorganisms8060903. Microorganisms. 2020. PMID: 32549402 Free PMC article.
-
An evolutionary model identifies the main evolutionary biases for the evolution of genome-replication profiles.Elife. 2021 May 20;10:e63542. doi: 10.7554/eLife.63542. Elife. 2021. PMID: 34013887 Free PMC article.
-
Integrated genomic analysis reveals key features of long undecoded transcript isoform-based gene repression.Mol Cell. 2021 May 20;81(10):2231-2245.e11. doi: 10.1016/j.molcel.2021.03.013. Epub 2021 Apr 6. Mol Cell. 2021. PMID: 33826921 Free PMC article.
-
The Genome Sequence of the Jean-Talon Strain, an Archeological Beer Yeast from Québec, Reveals Traces of Adaptation to Specific Brewing Conditions.G3 (Bethesda). 2020 Sep 2;10(9):3087-3097. doi: 10.1534/g3.120.401149. G3 (Bethesda). 2020. PMID: 32605927 Free PMC article.
-
Adaptation of the yeast gene knockout collection is near-perfectly predicted by fitness and diminishing return epistasis.G3 (Bethesda). 2022 Nov 4;12(11):jkac240. doi: 10.1093/g3journal/jkac240. G3 (Bethesda). 2022. PMID: 36083011 Free PMC article.
References
-
- Cao J, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
