

Localized TWIST1 and TWIST2 basic domain substitutions cause four distinct human diseases that can be modeled in Caenorhabditis elegans
- PMID: 28369379
- PMCID: PMC5438873
- DOI: 10.1093/hmg/ddx107
Localized TWIST1 and TWIST2 basic domain substitutions cause four distinct human diseases that can be modeled in Caenorhabditis elegans
Abstract
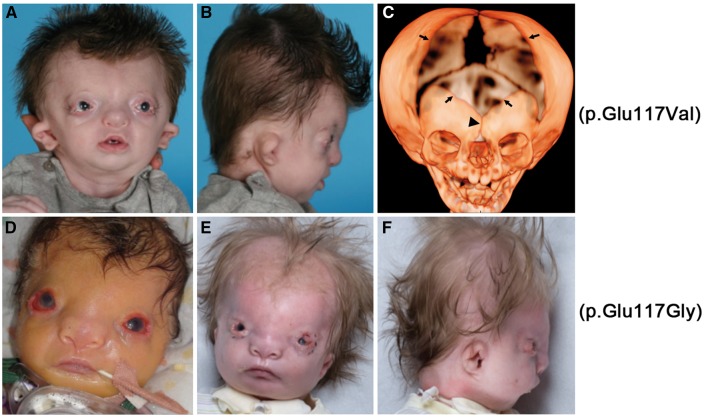
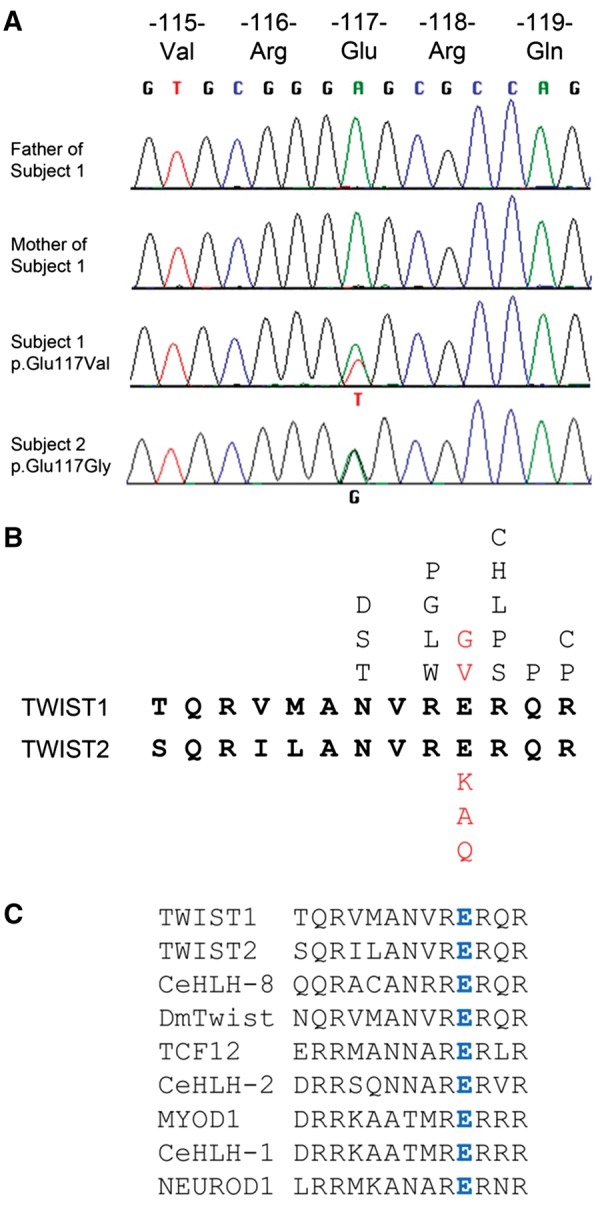
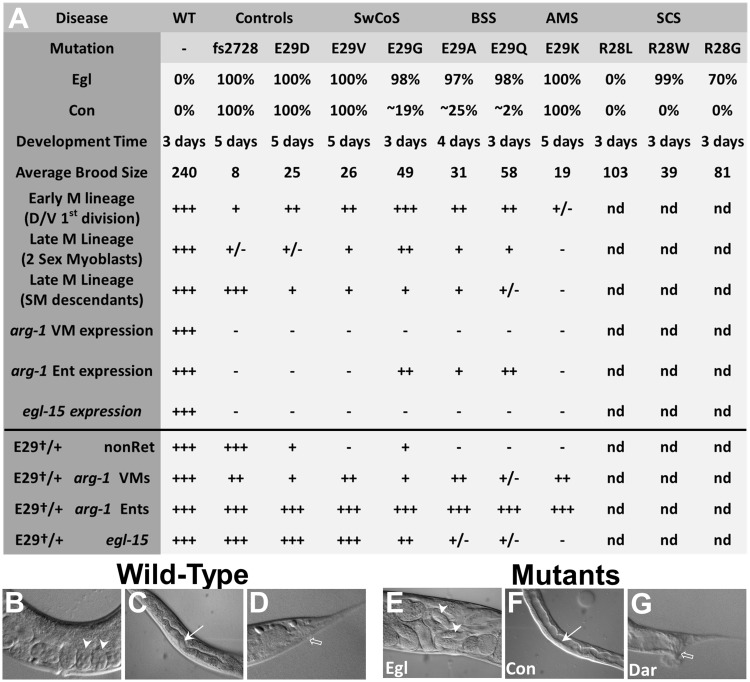
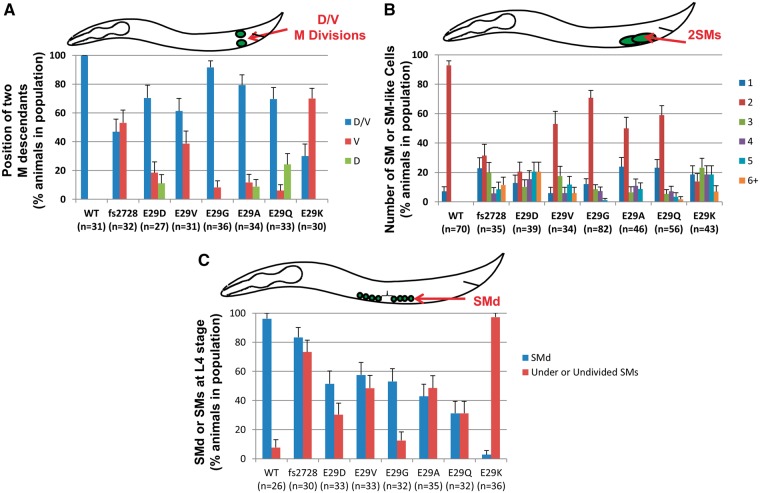
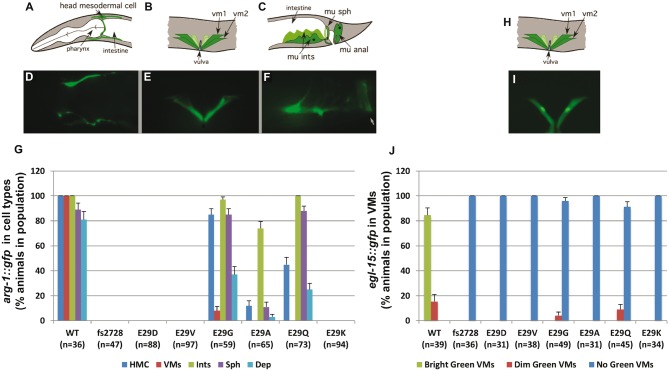
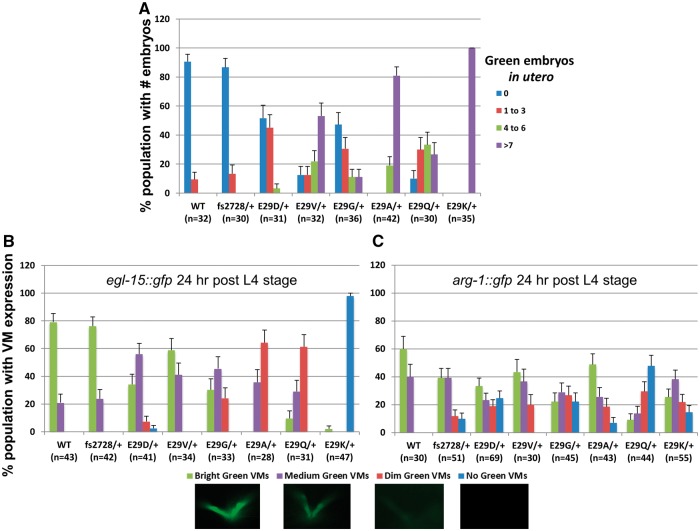
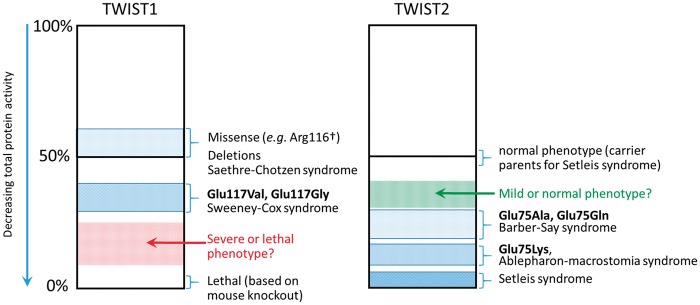
Twist transcription factors, members of the basic helix-loop-helix family, play crucial roles in mesoderm development in all animals. Humans have two paralogous genes, TWIST1 and TWIST2, and mutations in each gene have been identified in specific craniofacial disorders. Here, we describe a new clinical entity, Sweeney-Cox syndrome, associated with distinct de novo amino acid substitutions (p.Glu117Val and p.Glu117Gly) at a highly conserved glutamic acid residue located in the basic DNA binding domain of TWIST1, in two subjects with frontonasal dysplasia and additional malformations. Although about one hundred different TWIST1 mutations have been reported in patients with the dominant haploinsufficiency Saethre-Chotzen syndrome (typically associated with craniosynostosis), substitutions uniquely affecting the Glu117 codon were not observed previously. Recently, subjects with Barber-Say and Ablepharon-Macrostomia syndromes were found to harbor heterozygous missense substitutions in the paralogous glutamic acid residue in TWIST2 (p.Glu75Ala, p.Glu75Gln and p.Glu75Lys). To study systematically the effects of these substitutions in individual cells of the developing mesoderm, we engineered all five disease-associated alleles into the equivalent Glu29 residue encoded by hlh-8, the single Twist homolog present in Caenorhabditis elegans. This allelic series revealed that different substitutions exhibit graded severity, in terms of both gene expression and cellular phenotype, which we incorporate into a model explaining the various human disease phenotypes. The genetic analysis favors a predominantly dominant-negative mechanism for the action of amino acid substitutions at this highly conserved glutamic acid residue and illustrates the value of systematic mutagenesis of C. elegans for focused investigation of human disease processes.
Published by Oxford University Press 2017. This work is written by US Government employees and is in the public domain in the US.
Figures
Similar articles
-
Ablepharon and craniosynostosis in a patient with a localized TWIST1 basic domain substitution.Am J Med Genet A. 2018 Dec;176(12):2777-2780. doi: 10.1002/ajmg.a.40525. Epub 2018 Nov 18. Am J Med Genet A. 2018. PMID: 30450715
-
Recurrent Mutations in the Basic Domain of TWIST2 Cause Ablepharon Macrostomia and Barber-Say Syndromes.Am J Hum Genet. 2015 Jul 2;97(1):99-110. doi: 10.1016/j.ajhg.2015.05.017. Epub 2015 Jun 25. Am J Hum Genet. 2015. PMID: 26119818 Free PMC article.
-
A Twist-Box domain of the C. elegans Twist homolog, HLH-8, plays a complex role in transcriptional regulation.Genetics. 2023 Aug 9;224(4):iyad066. doi: 10.1093/genetics/iyad066. Genetics. 2023. PMID: 37067863 Free PMC article.
-
Phosphoregulation of Twist1 provides a mechanism of cell fate control.Curr Med Chem. 2008;15(25):2641-7. doi: 10.2174/092986708785908987. Curr Med Chem. 2008. PMID: 18855684 Free PMC article. Review.
-
Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms.Cell Res. 2012 Jan;22(1):90-106. doi: 10.1038/cr.2011.144. Epub 2011 Aug 30. Cell Res. 2012. PMID: 21876555 Free PMC article. Review.
Cited by
-
Mechanisms of Regulation of the CHRDL1 Gene by the TWIST2 and ADD1/SREBP1c Transcription Factors.Genes (Basel). 2023 Aug 30;14(9):1733. doi: 10.3390/genes14091733. Genes (Basel). 2023. PMID: 37761873 Free PMC article.
-
Using Caenorhabditis elegans as a Model for Mechanistic Insights of Craniofacial Development.Methods Mol Biol. 2022;2403:1-18. doi: 10.1007/978-1-0716-1847-9_1. Methods Mol Biol. 2022. PMID: 34913112 Free PMC article.
-
New locus underlying auriculocondylar syndrome (ARCND): 430 kb duplication involving TWIST1 regulatory elements.J Med Genet. 2022 Sep;59(9):895-905. doi: 10.1136/jmedgenet-2021-107825. Epub 2021 Nov 8. J Med Genet. 2022. PMID: 34750192 Free PMC article.
-
DNA-guided transcription factor cooperativity shapes face and limb mesenchyme.Cell. 2024 Feb 1;187(3):692-711.e26. doi: 10.1016/j.cell.2023.12.032. Epub 2024 Jan 22. Cell. 2024. PMID: 38262408
-
Model organisms contribute to diagnosis and discovery in the undiagnosed diseases network: current state and a future vision.Orphanet J Rare Dis. 2021 May 7;16(1):206. doi: 10.1186/s13023-021-01839-9. Orphanet J Rare Dis. 2021. PMID: 33962631 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
