

Human Cytomegalovirus MicroRNAs miR-US5-1 and miR-UL112-3p Block Proinflammatory Cytokine Production in Response to NF-κB-Activating Factors through Direct Downregulation of IKKα and IKKβ
- PMID: 28270578
- PMCID: PMC5340867
- DOI: 10.1128/mBio.00109-17
Human Cytomegalovirus MicroRNAs miR-US5-1 and miR-UL112-3p Block Proinflammatory Cytokine Production in Response to NF-κB-Activating Factors through Direct Downregulation of IKKα and IKKβ
Abstract
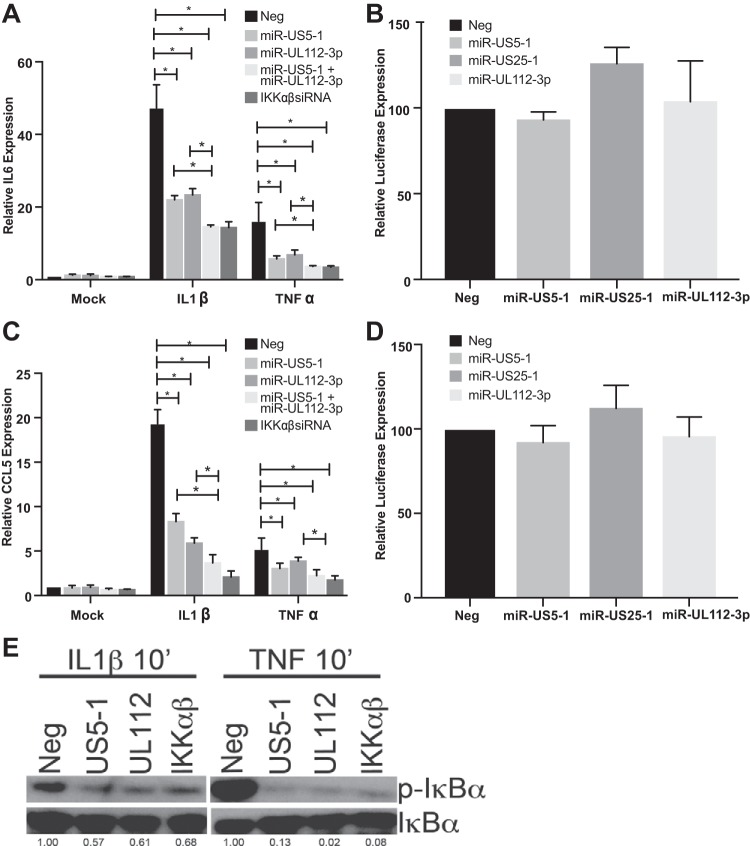
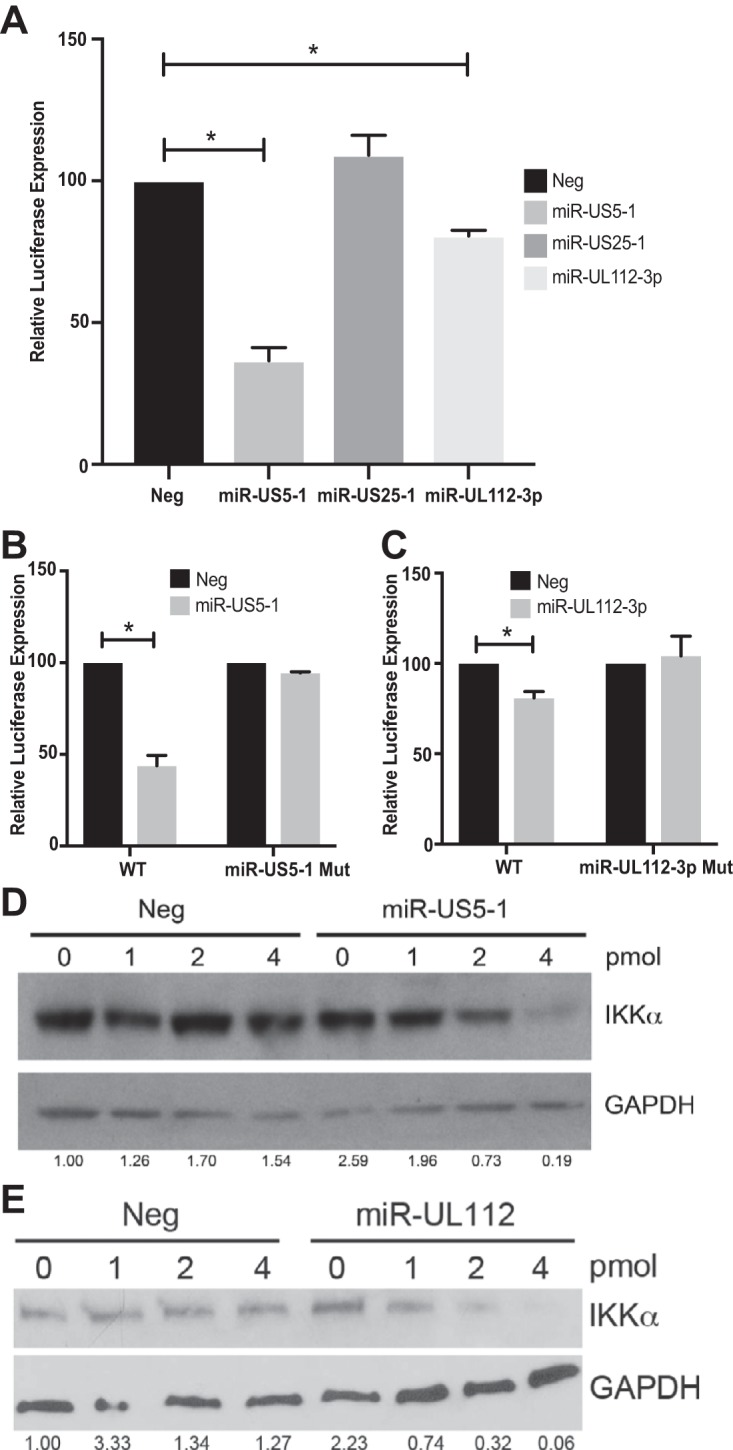
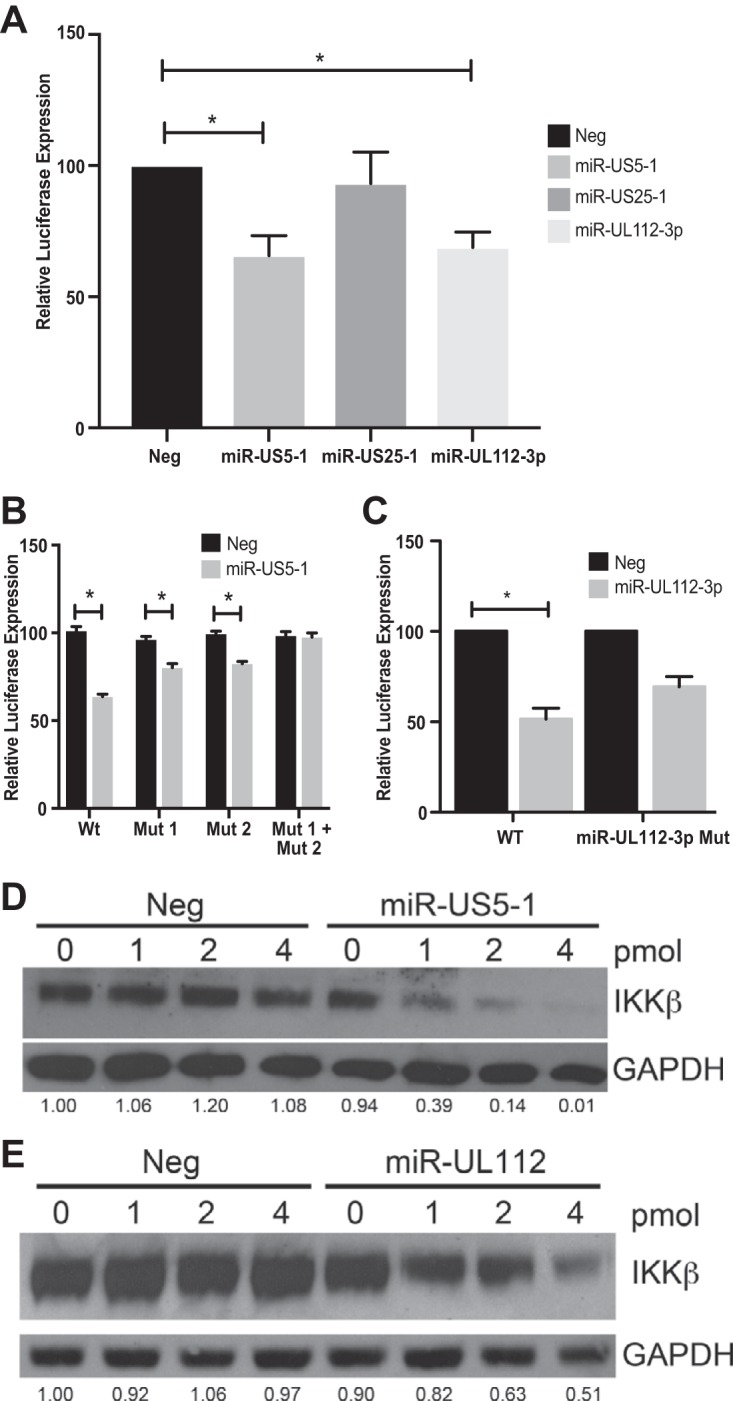
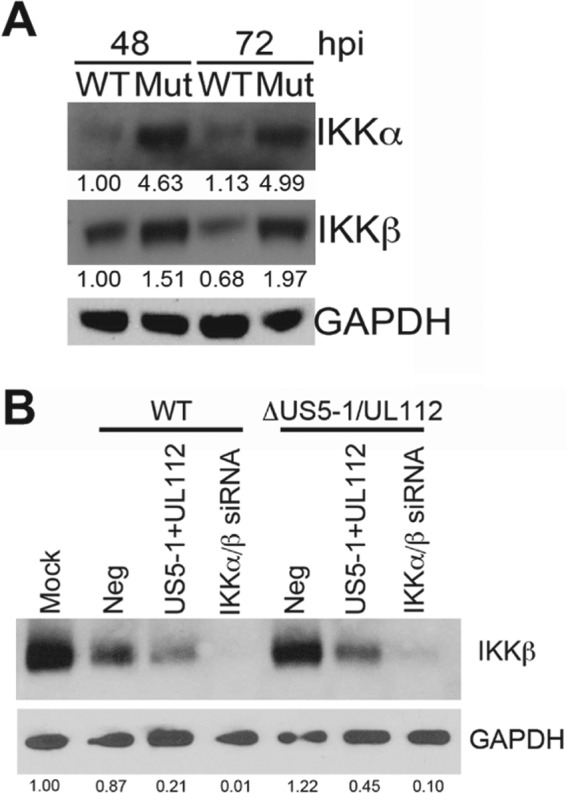
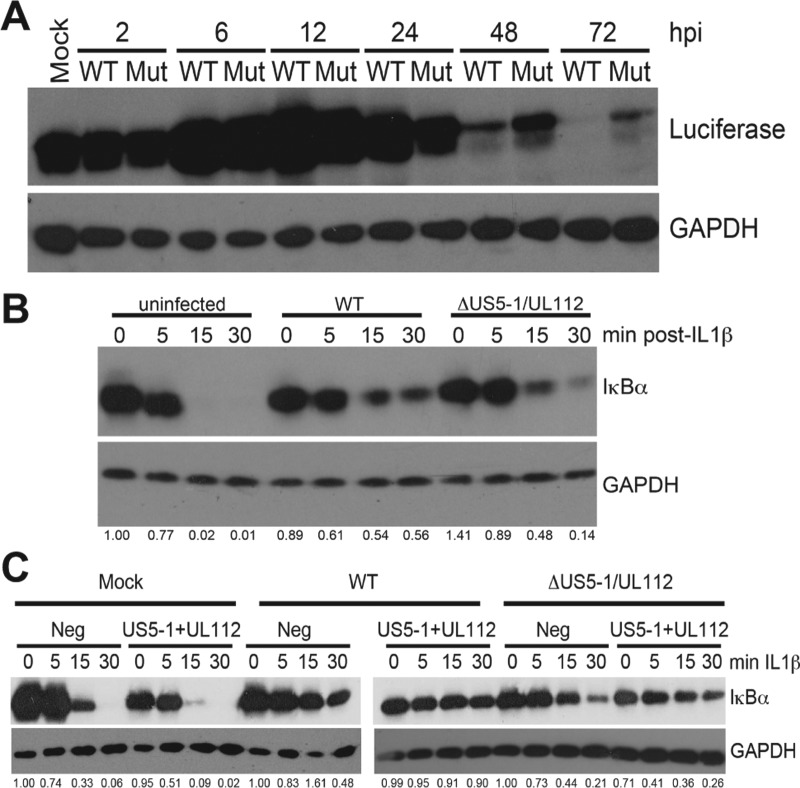
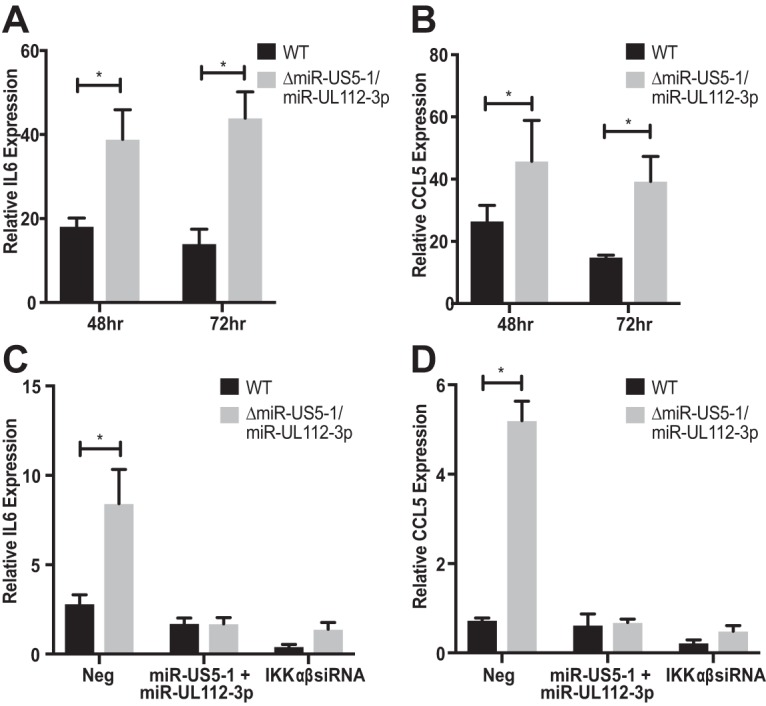
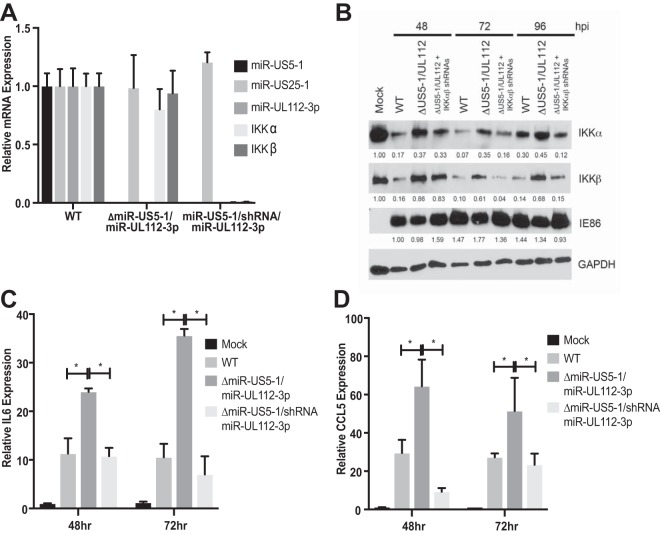
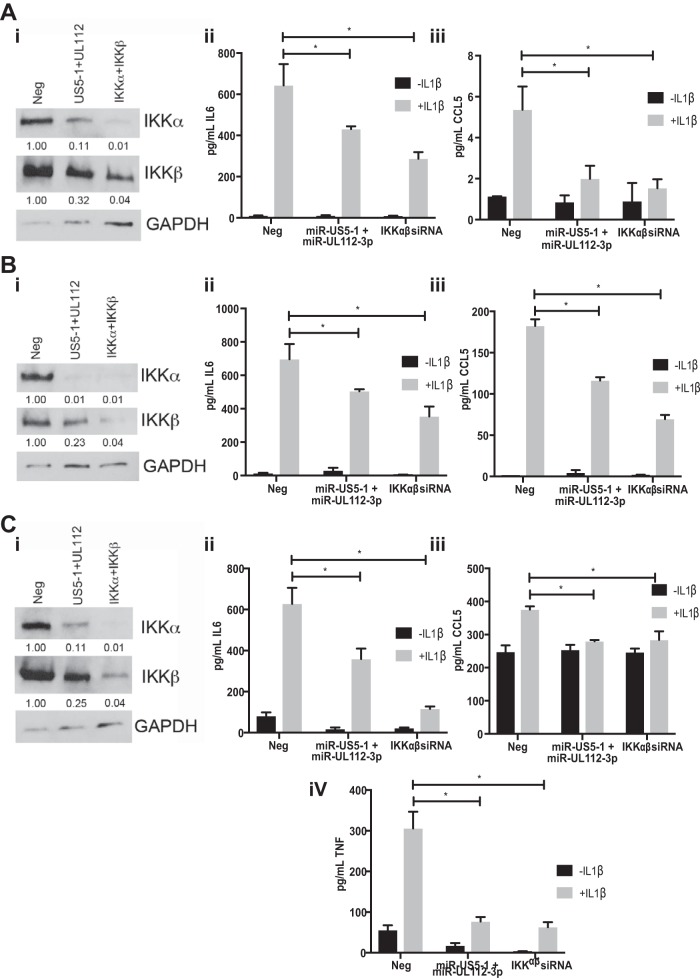
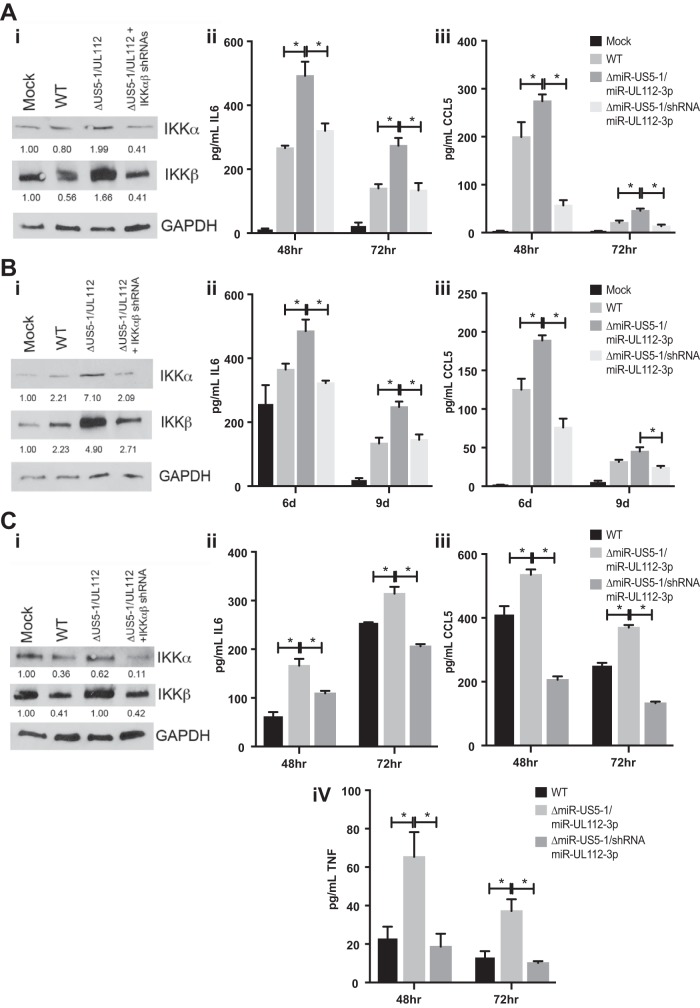
Emerging evidence indicates that human cytomegalovirus (HCMV) manipulates host cell signaling pathways using both proteins and noncoding RNAs. Several studies have shown that HCMV induces NF-κB signaling early in infection, resulting in the induction of antiviral proinflammatory cytokines with a subsequent reduction of these cytokines late in infection. The mechanism for late cytokine reduction is unknown. In this study, we show that HCMV microRNAs (miRNAs) miR-US5-1 and miR-UL112-3p target the IκB kinase (IKK) complex components IKKα and IKKβ to limit production of proinflammatory cytokines in response to interleukin 1β (IL-1β) and tumor necrosis factor alpha (TNF-α). Transfection of miR-UL112-3p and miR-US5-1 mimics reduced endogenous IKKα and IKKβ protein levels, and site-directed mutagenesis of the 3' untranslated regions (UTRs) identified the binding sites for each miRNA. Infection with mutant viruses lacking these miRNAs resulted in increased levels of IKKα and IKKβ proteins, an impaired ability to control NF-κB signaling at late times of lytic infection, and increased production of proinflammatory cytokines compared to wild-type virus in cell types relevant to HCMV infection in vivo These phenotypes were rescued by preexpression of miR-US5-1 and miR-UL112-3p in infected cells or by a miR-US5-1/miR-UL112-3p double mutant virus that expresses short hairpin RNAs (shRNAs) targeting IKKα and IKKβ, demonstrating the gene specificity of the miRNAs. These observations describe a mechanism through which HCMV miRNAs expressed late in the infectious cycle downregulate proinflammatory cytokine production to create a cellular proviral environment.IMPORTANCE Human cytomegalovirus (HCMV) is a significant cause of morbidity and mortality in transplant recipients and causes hearing loss and mental retardation when acquired congenitally. Initial events during HCMV infection result in the activation of NF-κB signaling, which culminates in the production of IL-6, CCL5, and TNF-α. Several viruses have developed mechanisms to block the antiviral effects of these cytokines. We show here that two HCMV miRNAs, miR-US5-1 and miR-UL112-3p, specifically downregulate IKKα and IKKβ signaling factors necessary to propagate NF-κB signaling and subsequent IL-6, CCL5, and TNF-α production. Regulation of these proinflammatory cytokines during lytic infection and during latency is critical to viral survival in the host.
Copyright © 2017 Hancock et al.
Figures
Comment in
-
New Mechanism by Which Human Cytomegalovirus MicroRNAs Negate the Proinflammatory Response to Infection.mBio. 2017 Apr 18;8(2):e00505-17. doi: 10.1128/mBio.00505-17. mBio. 2017. PMID: 28420741 Free PMC article.
Similar articles
-
New Mechanism by Which Human Cytomegalovirus MicroRNAs Negate the Proinflammatory Response to Infection.mBio. 2017 Apr 18;8(2):e00505-17. doi: 10.1128/mBio.00505-17. mBio. 2017. PMID: 28420741 Free PMC article.
-
Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway.PLoS Pathog. 2015 May 8;11(5):e1004881. doi: 10.1371/journal.ppat.1004881. eCollection 2015 May. PLoS Pathog. 2015. PMID: 25955717 Free PMC article.
-
UL26 Attenuates IKKβ-Mediated Induction of Interferon-Stimulated Gene (ISG) Expression and Enhanced Protein ISGylation during Human Cytomegalovirus Infection.J Virol. 2019 Nov 13;93(23):e01052-19. doi: 10.1128/JVI.01052-19. Print 2019 Dec 1. J Virol. 2019. PMID: 31534044 Free PMC article.
-
Regulation and function of IKK and IKK-related kinases.Sci STKE. 2006 Oct 17;2006(357):re13. doi: 10.1126/stke.3572006re13. Sci STKE. 2006. PMID: 17047224 Review.
-
Regulation of the MIR155 host gene in physiological and pathological processes.Gene. 2013 Dec 10;532(1):1-12. doi: 10.1016/j.gene.2012.12.009. Epub 2012 Dec 14. Gene. 2013. PMID: 23246696 Review.
Cited by
-
Control of Cytokines in Latent Cytomegalovirus Infection.Pathogens. 2020 Oct 21;9(10):858. doi: 10.3390/pathogens9100858. Pathogens. 2020. PMID: 33096622 Free PMC article. Review.
-
Epstein-Barr virus microRNAs regulate B cell receptor signal transduction and lytic reactivation.PLoS Pathog. 2019 Jan 7;15(1):e1007535. doi: 10.1371/journal.ppat.1007535. eCollection 2019 Jan. PLoS Pathog. 2019. PMID: 30615681 Free PMC article.
-
A Prominent Role of the Human Cytomegalovirus UL8 Glycoprotein in Restraining Proinflammatory Cytokine Production by Myeloid Cells at Late Times during Infection.J Virol. 2018 Apr 13;92(9):e02229-17. doi: 10.1128/JVI.02229-17. Print 2018 May 1. J Virol. 2018. PMID: 29467314 Free PMC article.
-
Viral miRNA regulation of host gene expression.Semin Cell Dev Biol. 2023 Sep 15;146:2-19. doi: 10.1016/j.semcdb.2022.11.007. Epub 2022 Nov 30. Semin Cell Dev Biol. 2023. PMID: 36463091 Free PMC article. Review.
-
Human Cytomegalovirus miRNAs Regulate TGF-β to Mediate Myelosuppression while Maintaining Viral Latency in CD34+ Hematopoietic Progenitor Cells.Cell Host Microbe. 2020 Jan 8;27(1):104-114.e4. doi: 10.1016/j.chom.2019.11.013. Epub 2019 Dec 19. Cell Host Microbe. 2020. PMID: 31866424 Free PMC article.
References
-
- Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. 1995. Signal-induced site-specific phosphorylation targets IkappaBalpha to the ubiquitin-proteasome pathway. Genes Dev 9:1586–1597. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
