

Interaction of the polyglutamine protein ataxin-3 with Rad23 regulates toxicity in Drosophila models of Spinocerebellar Ataxia Type 3
- PMID: 28158474
- PMCID: PMC6075452
- DOI: 10.1093/hmg/ddx039
Interaction of the polyglutamine protein ataxin-3 with Rad23 regulates toxicity in Drosophila models of Spinocerebellar Ataxia Type 3
Abstract
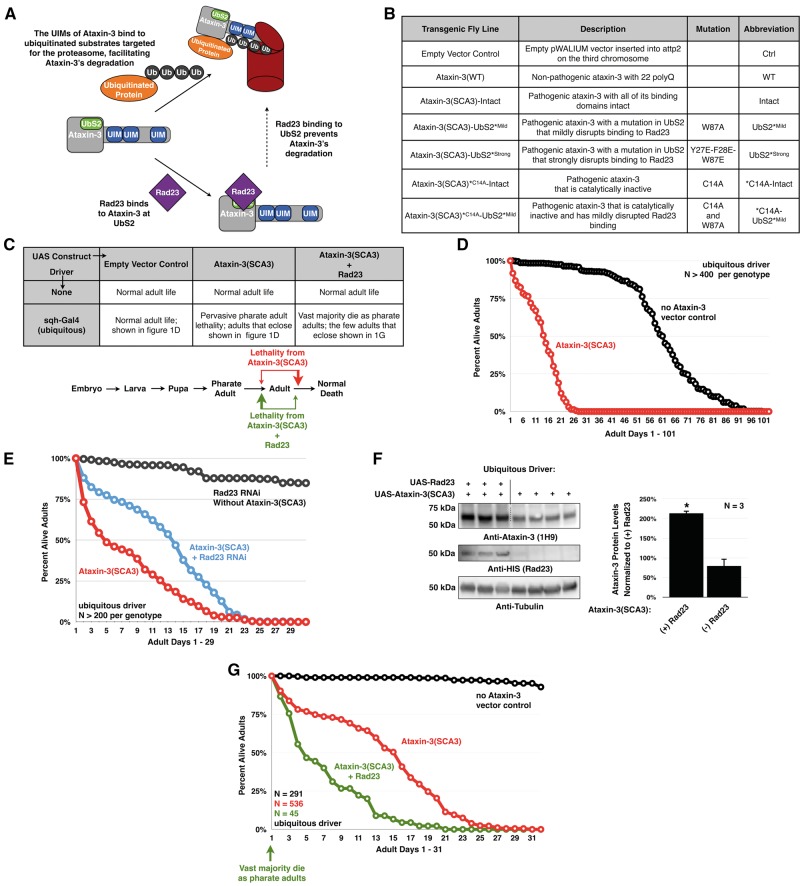
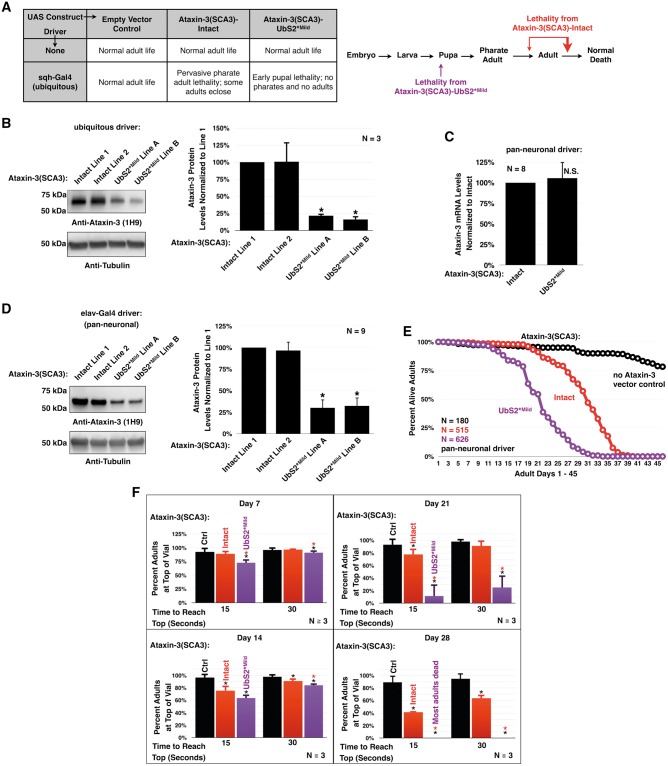
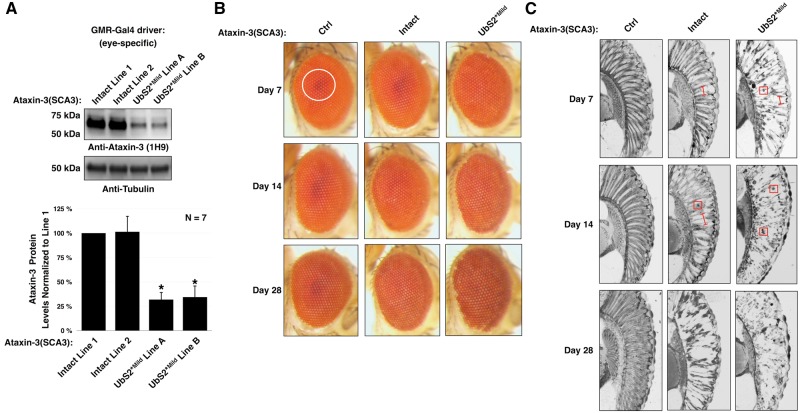
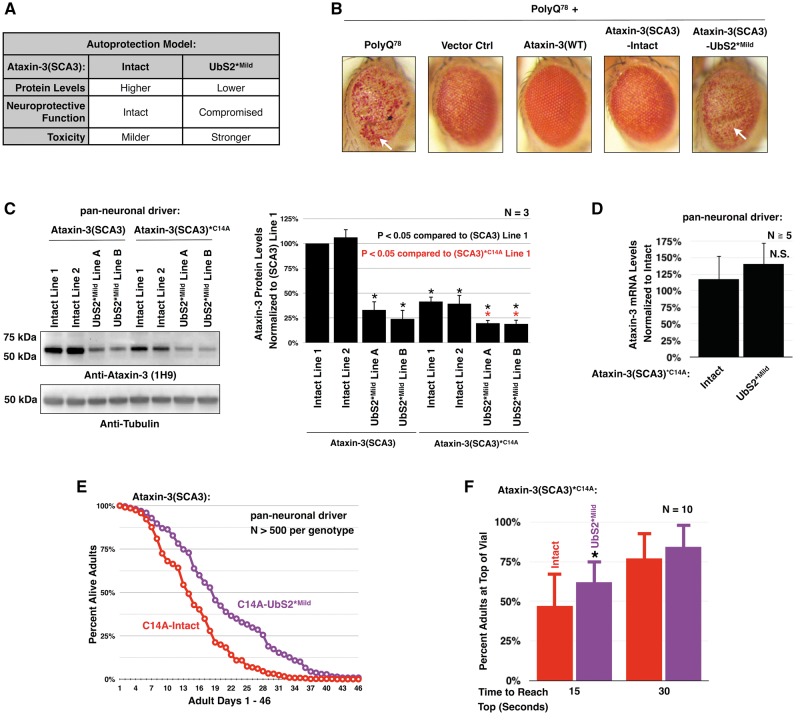
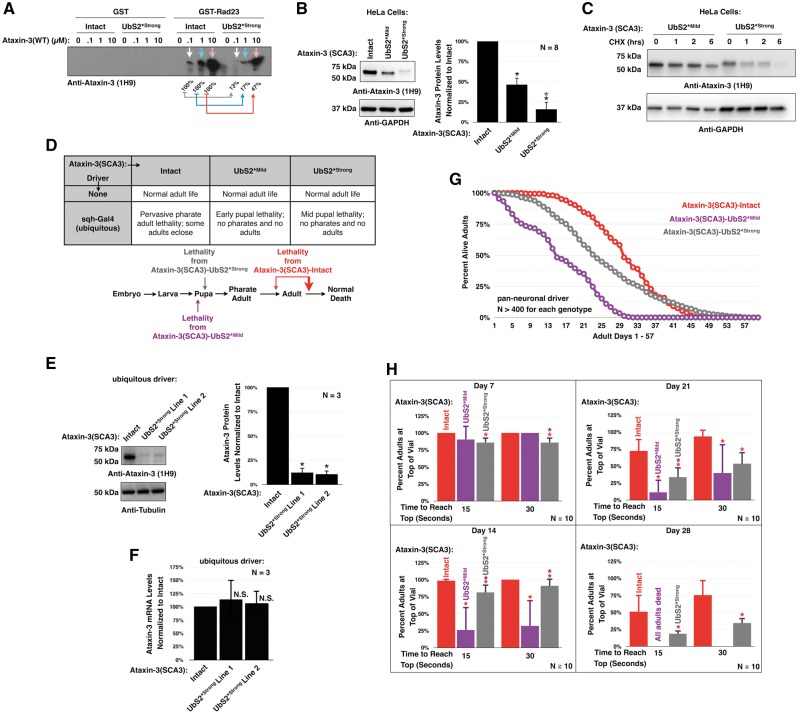
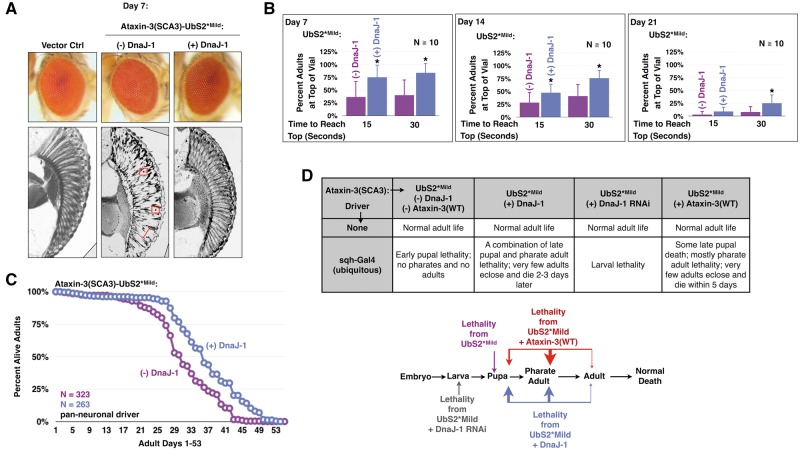
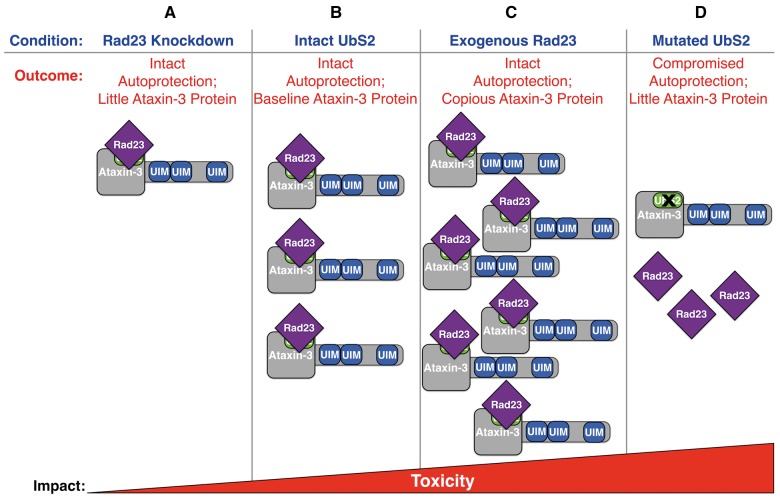
Polyglutamine (polyQ) repeat expansion in the deubiquitinase ataxin-3 causes neurodegeneration in Spinocerebellar Ataxia Type 3 (SCA3), one of nine inherited, incurable diseases caused by similar mutations. Ataxin-3's degradation is inhibited by its binding to the proteasome shuttle Rad23 through ubiquitin-binding site 2 (UbS2). Disrupting this interaction decreases levels of ataxin-3. Since reducing levels of polyQ proteins can decrease their toxicity, we tested whether genetically modulating the ataxin-3-Rad23 interaction regulates its toxicity in Drosophila. We found that exogenous Rad23 increases the toxicity of pathogenic ataxin-3, coincident with increased levels of the disease protein. Conversely, reducing Rad23 levels alleviates toxicity in this SCA3 model. Unexpectedly, pathogenic ataxin-3 with a mutated Rad23-binding site at UbS2, despite being present at markedly lower levels, proved to be more pathogenic than a disease-causing counterpart with intact UbS2. Additional studies established that the increased toxicity upon mutating UbS2 stems from disrupting the autoprotective role that pathogenic ataxin-3 has against itself, which depends on the co-chaperone, DnaJ-1. Our data reveal a previously unrecognized balance between pathogenic and potentially therapeutic properties of the ataxin-3-Rad23 interaction; they highlight this interaction as critical for the toxicity of the SCA3 protein, and emphasize the importance of considering protein context when pursuing suppressive avenues.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23.Nat Commun. 2014 Aug 21;5:4638. doi: 10.1038/ncomms5638. Nat Commun. 2014. PMID: 25144244 Free PMC article.
-
The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster.Neurobiol Dis. 2015 Oct;82:12-21. doi: 10.1016/j.nbd.2015.05.010. Epub 2015 May 22. Neurobiol Dis. 2015. PMID: 26007638 Free PMC article.
-
A fine balance between Prpf19 and Exoc7 in achieving degradation of aggregated protein and suppression of cell death in spinocerebellar ataxia type 3.Cell Death Dis. 2021 Feb 2;12(2):136. doi: 10.1038/s41419-021-03444-x. Cell Death Dis. 2021. PMID: 33542212 Free PMC article.
-
Toward therapeutic targets for SCA3: Insight into the role of Machado-Joseph disease protein ataxin-3 in misfolded proteins clearance.Prog Neurobiol. 2015 Sep;132:34-58. doi: 10.1016/j.pneurobio.2015.06.004. Epub 2015 Jun 27. Prog Neurobiol. 2015. PMID: 26123252 Review.
-
The deubiquitinase function of ataxin-3 and its role in the pathogenesis of Machado-Joseph disease and other diseases.Biochem J. 2024 Mar 20;481(6):461-480. doi: 10.1042/BCJ20240017. Biochem J. 2024. PMID: 38497605 Free PMC article. Review.
Cited by
-
Degron capability of the hydrophobic C-terminus of the polyglutamine disease protein, ataxin-3.J Neurosci Res. 2020 Oct;98(10):2096-2108. doi: 10.1002/jnr.24684. Epub 2020 Jul 9. J Neurosci Res. 2020. PMID: 32643791 Free PMC article.
-
Endurance exercise ameliorates phenotypes in Drosophila models of spinocerebellar ataxias.Elife. 2022 Feb 16;11:e75389. doi: 10.7554/eLife.75389. Elife. 2022. PMID: 35170431 Free PMC article.
-
Deubiquitinase USP7 contributes to the pathogenicity of spinal and bulbar muscular atrophy.J Clin Invest. 2021 Jan 4;131(1):e134565. doi: 10.1172/JCI134565. J Clin Invest. 2021. PMID: 33170804 Free PMC article.
-
Progressive degeneration in a new Drosophila model of Spinocerebellar Ataxia type 7.Res Sq [Preprint]. 2023 Nov 23:rs.3.rs-3592641. doi: 10.21203/rs.3.rs-3592641/v1. Res Sq. 2023. Update in: Sci Rep. 2024 Jun 21;14(1):14332. doi: 10.1038/s41598-024-65172-4. PMID: 38045332 Free PMC article. Updated. Preprint.
-
Ubiquitin signaling in neurodegenerative diseases: an autophagy and proteasome perspective.Cell Death Differ. 2021 Feb;28(2):439-454. doi: 10.1038/s41418-020-00667-x. Epub 2020 Nov 18. Cell Death Differ. 2021. PMID: 33208890 Free PMC article. Review.
References
-
- Todi S.V., Williams A., Paulson H. (2007) Waxman S.G. (ed.), In Molecular Neurology. Academic Press, London, in press., pp. 257–276.
-
- Matos C.A., de Macedo-Ribeiro S., Carvalho A.L. (2011) Polyglutamine diseases: The special case of ataxin-3 and Machado-Joseph disease. Prog. Neurobiol., 95, 26–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
