

Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species
- PMID: 28148785
- PMCID: PMC5375695
- DOI: 10.1128/JVI.02381-16
Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species
Abstract
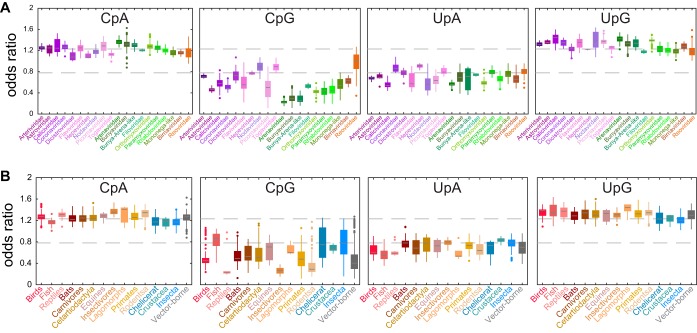
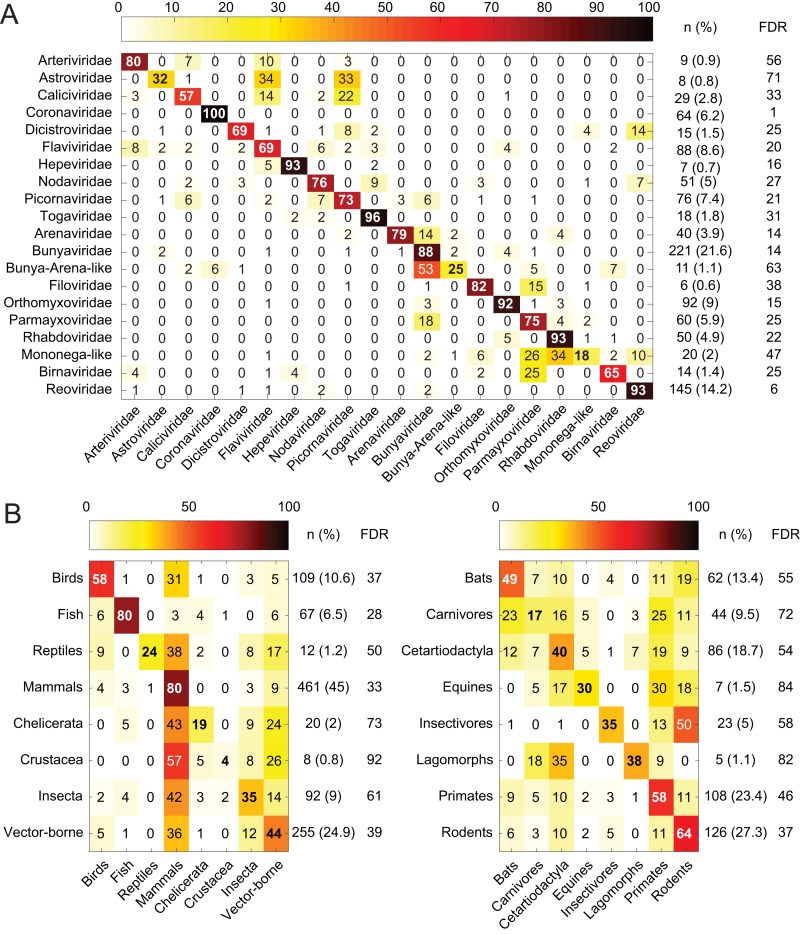
Viruses use the cellular machinery of their hosts for replication. It has therefore been proposed that the nucleotide and dinucleotide compositions of viruses should match those of their host species. If this is upheld, it may then be possible to use dinucleotide composition to predict the true host species of viruses sampled in metagenomic surveys. However, it is also clear that different taxonomic groups of viruses tend to have distinctive patterns of dinucleotide composition that may be independent of host species. To determine the relative strength of the effect of host versus virus family in shaping dinucleotide composition, we performed a comparative analysis of 20 RNA virus families from 15 host groupings, spanning two animal phyla and more than 900 virus species. In particular, we determined the odds ratios for the 16 possible dinucleotides and performed a discriminant analysis to evaluate the capability of virus dinucleotide composition to predict the correct virus family or host taxon from which it was isolated. Notably, while 81% of the data analyzed here were predicted to the correct virus family, only 62% of these data were predicted to their correct subphylum/class host and a mere 32% to their correct mammalian order. Similarly, dinucleotide composition has a weak predictive power for different hosts within individual virus families. We therefore conclude that dinucleotide composition is generally uniform within a virus family but less well reflects that of its host species. This has obvious implications for attempts to accurately predict host species from virus genome sequences alone.IMPORTANCE Determining the processes that shape virus genomes is central to understanding virus evolution and emergence. One question of particular importance is why nucleotide and dinucleotide frequencies differ so markedly between viruses. In particular, it is currently unclear whether host species or virus family has the biggest impact on dinucleotide frequencies and whether dinucleotide composition can be used to accurately predict host species. Using a comparative analysis, we show that dinucleotide composition has a strong phylogenetic association across different RNA virus families, such that dinucleotide composition can predict the family from which a virus sequence has been isolated. Conversely, dinucleotide composition has a poorer predictive power for the different host species within a virus family and across different virus families, indicating that the host has a relatively small impact on the dinucleotide composition of a virus genome.
Keywords: dinucleotide bias; evolution.
Copyright © 2017 American Society for Microbiology.
Figures


Similar articles
-
Patterns of evolution and host gene mimicry in influenza and other RNA viruses.PLoS Pathog. 2008 Jun 6;4(6):e1000079. doi: 10.1371/journal.ppat.1000079. PLoS Pathog. 2008. PMID: 18535658 Free PMC article.
-
Does the Zinc Finger Antiviral Protein (ZAP) Shape the Evolution of Herpesvirus Genomes?Viruses. 2021 Sep 17;13(9):1857. doi: 10.3390/v13091857. Viruses. 2021. PMID: 34578438 Free PMC article. Review.
-
Compositional biases and evolution of the largest plant RNA virus order Patatavirales.Int J Biol Macromol. 2023 Jun 15;240:124403. doi: 10.1016/j.ijbiomac.2023.124403. Epub 2023 Apr 18. Int J Biol Macromol. 2023. PMID: 37076075
-
Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses?J Virol. 1994 May;68(5):2889-97. doi: 10.1128/JVI.68.5.2889-2897.1994. J Virol. 1994. PMID: 8151759 Free PMC article.
-
Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences.Crit Rev Biochem Mol Biol. 1993;28(5):375-430. doi: 10.3109/10409239309078440. Crit Rev Biochem Mol Biol. 1993. PMID: 8269709 Review.
Cited by
-
Depletion of CpG dinucleotides in bacterial genomes may represent an adaptation to high temperatures.NAR Genom Bioinform. 2024 Jul 27;6(3):lqae088. doi: 10.1093/nargab/lqae088. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 39071851 Free PMC article.
-
Dengue virus preferentially uses human and mosquito non-optimal codons.Mol Syst Biol. 2024 Oct;20(10):1085-1108. doi: 10.1038/s44320-024-00052-7. Epub 2024 Jul 22. Mol Syst Biol. 2024. PMID: 39039212 Free PMC article.
-
Xinyang flavivirus, from Haemaphysalis flava ticks in Henan Province, China, defines a basal, likely tick-only Orthoflavivirus clade.J Gen Virol. 2024 May;105(5):001991. doi: 10.1099/jgv.0.001991. J Gen Virol. 2024. PMID: 38809251 Free PMC article.
-
Molecular Characterization and Genome Mechanical Features of Two Newly Isolated Polyvalent Bacteriophages Infecting Pseudomonas syringae pv. garcae.Genes (Basel). 2024 Jan 18;15(1):113. doi: 10.3390/genes15010113. Genes (Basel). 2024. PMID: 38255005 Free PMC article.
-
A novel tamanavirus (Flaviviridae) of the European common frog (Rana temporaria) from the UK.J Gen Virol. 2023 Dec;104(12):001927. doi: 10.1099/jgv.0.001927. J Gen Virol. 2023. PMID: 38059479 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
