

Synthetic lethal mutations in the cyclin A interface of human cytomegalovirus
- PMID: 28129404
- PMCID: PMC5298330
- DOI: 10.1371/journal.ppat.1006193
Synthetic lethal mutations in the cyclin A interface of human cytomegalovirus
Abstract
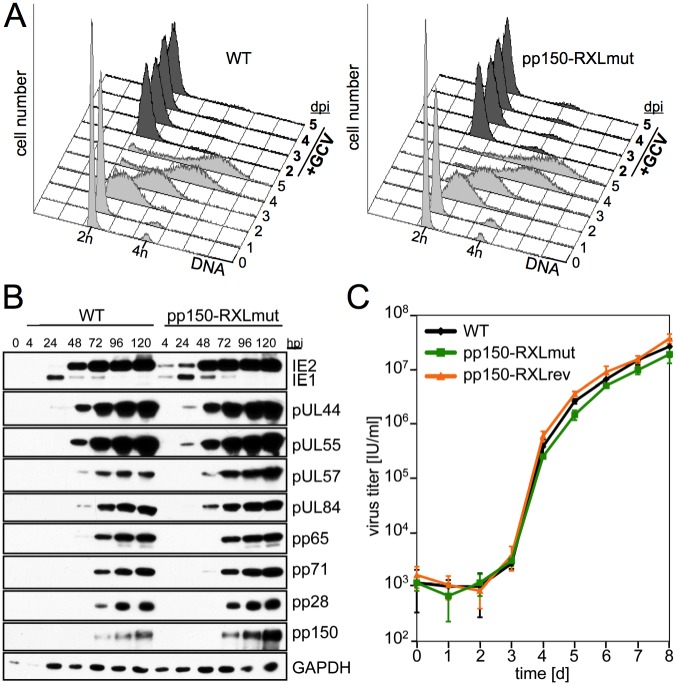
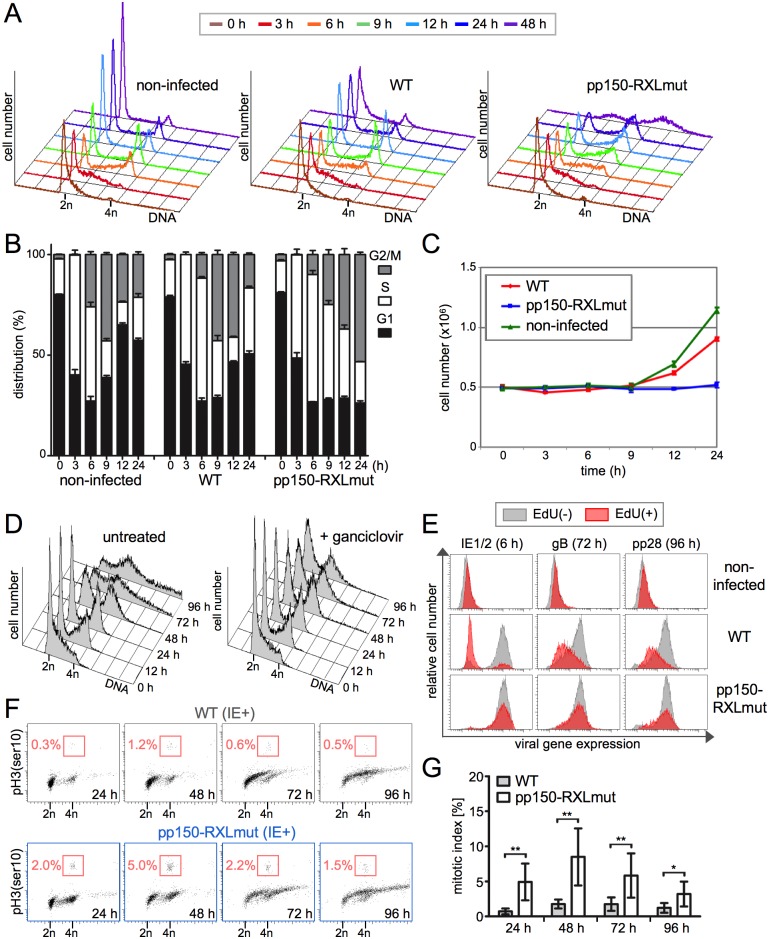
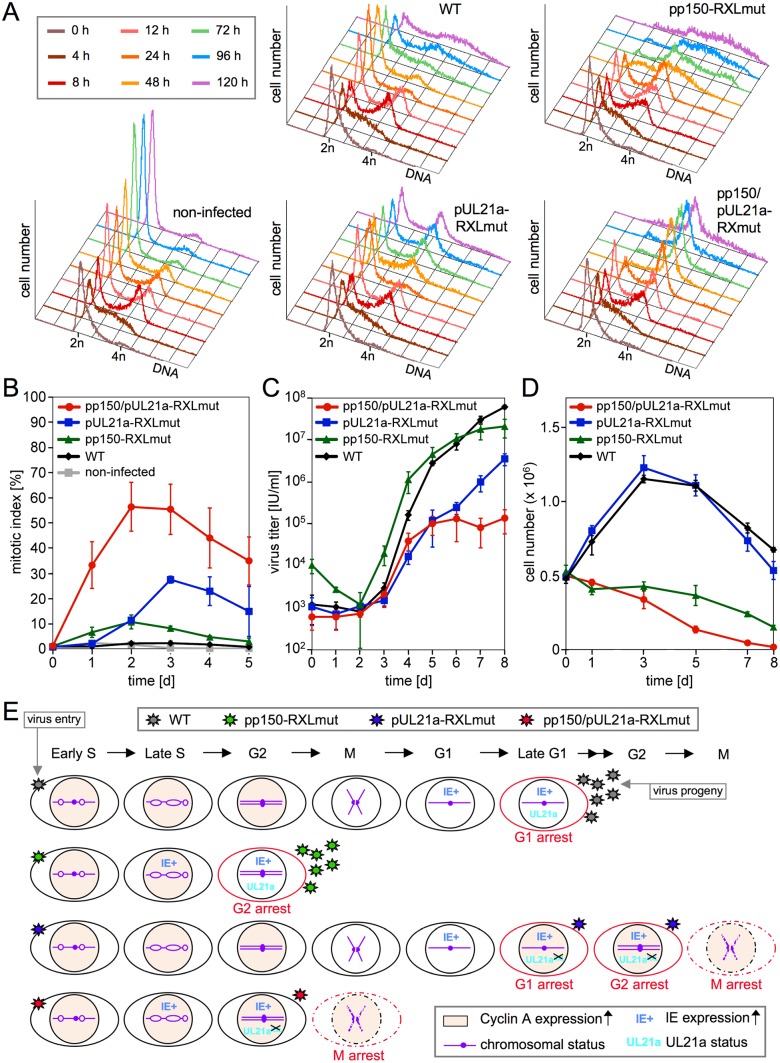
Generally, the antagonism between host restriction factors and viral countermeasures decides on cellular permissiveness or resistance to virus infection. Human cytomegalovirus (HCMV) has evolved an additional level of self-imposed restriction by the viral tegument protein pp150. Depending on a cyclin A-binding motif, pp150 prevents the onset of viral gene expression in the S/G2 cell cycle phase of otherwise fully permissive cells. Here we address the physiological relevance of this restriction during productive HCMV infection by employing a cyclin A-binding deficient pp150 mutant virus. One consequence of unrestricted viral gene expression in S/G2 was the induction of a G2/M arrest. G2-arrested but not mitotic cells supported viral replication. Cyclin A destabilization by the viral gene product pUL21a was required to maintain the virus-permissive G2-arrest. An HCMV double-point mutant where both pp150 and pUL21a are disabled in cyclin A interaction forced mitotic entry of the majority of infected cells, with a severe negative impact on cell viability and virus growth. Thus, pp150 and pUL21a functionally cooperate, together building a cell cycle synchronization strategy of cyclin A targeting and avoidance that is essential for productive HCMV infection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Human cytomegalovirus tegument protein pp150 acts as a cyclin A2-CDK-dependent sensor of the host cell cycle and differentiation state.Proc Natl Acad Sci U S A. 2013 Oct 22;110(43):17510-5. doi: 10.1073/pnas.1312235110. Epub 2013 Oct 7. Proc Natl Acad Sci U S A. 2013. PMID: 24101496 Free PMC article.
-
The smallest capsid protein mediates binding of the essential tegument protein pp150 to stabilize DNA-containing capsids in human cytomegalovirus.PLoS Pathog. 2013 Aug;9(8):e1003525. doi: 10.1371/journal.ppat.1003525. Epub 2013 Aug 15. PLoS Pathog. 2013. PMID: 23966856 Free PMC article.
-
PUL21a-Cyclin A2 interaction is required to protect human cytomegalovirus-infected cells from the deleterious consequences of mitotic entry.PLoS Pathog. 2014 Nov 13;10(10):e1004514. doi: 10.1371/journal.ppat.1004514. eCollection 2014 Oct. PLoS Pathog. 2014. PMID: 25393019 Free PMC article.
-
Human cytomegalovirus riding the cell cycle.Med Microbiol Immunol. 2015 Jun;204(3):409-19. doi: 10.1007/s00430-015-0396-z. Epub 2015 Mar 17. Med Microbiol Immunol. 2015. PMID: 25776080 Review.
-
HCMV infection: modulating the cell cycle and cell death.Int Rev Immunol. 2004 Jan-Apr;23(1-2):113-39. doi: 10.1080/08830180490265565. Int Rev Immunol. 2004. PMID: 14690857 Review.
Cited by
-
Human cytomegalovirus glycoprotein B variants affect viral entry, cell fusion, and genome stability.Proc Natl Acad Sci U S A. 2019 Sep 3;116(36):18021-18030. doi: 10.1073/pnas.1907447116. Epub 2019 Aug 19. Proc Natl Acad Sci U S A. 2019. PMID: 31427511 Free PMC article.
-
Emerging Mechanisms of G1/S Cell Cycle Control by Human and Mouse Cytomegaloviruses.mBio. 2021 Dec 21;12(6):e0293421. doi: 10.1128/mBio.02934-21. Epub 2021 Dec 14. mBio. 2021. PMID: 34903047 Free PMC article. Review.
-
Human Cytomegalovirus Genomes Survive Mitosis via the IE19 Chromatin-Tethering Domain.mBio. 2020 Sep 29;11(5):e02410-20. doi: 10.1128/mBio.02410-20. mBio. 2020. PMID: 32994332 Free PMC article.
-
Human Cytomegalovirus Primary Infection and Reactivation: Insights From Virion-Carried Molecules.Front Microbiol. 2020 Jul 14;11:1511. doi: 10.3389/fmicb.2020.01511. eCollection 2020. Front Microbiol. 2020. PMID: 32765441 Free PMC article. Review.
-
Triple lysine and nucleosome-binding motifs of the viral IE19 protein are required for human cytomegalovirus S-phase infections.mBio. 2024 Jun 12;15(6):e0016224. doi: 10.1128/mbio.00162-24. Epub 2024 May 2. mBio. 2024. PMID: 38695580 Free PMC article.
References
-
- Morgan DO. The Cell Cycle: Principles of Control. London: New Science Press; 2007.
-
- Mittnacht S, Boshoff C. Viral cyclins. Rev Med Virol. 2000;10: 175–184. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
