

Human AML-iPSCs Reacquire Leukemic Properties after Differentiation and Model Clonal Variation of Disease
- PMID: 28089908
- PMCID: PMC5508733
- DOI: 10.1016/j.stem.2016.11.018
Human AML-iPSCs Reacquire Leukemic Properties after Differentiation and Model Clonal Variation of Disease
Abstract
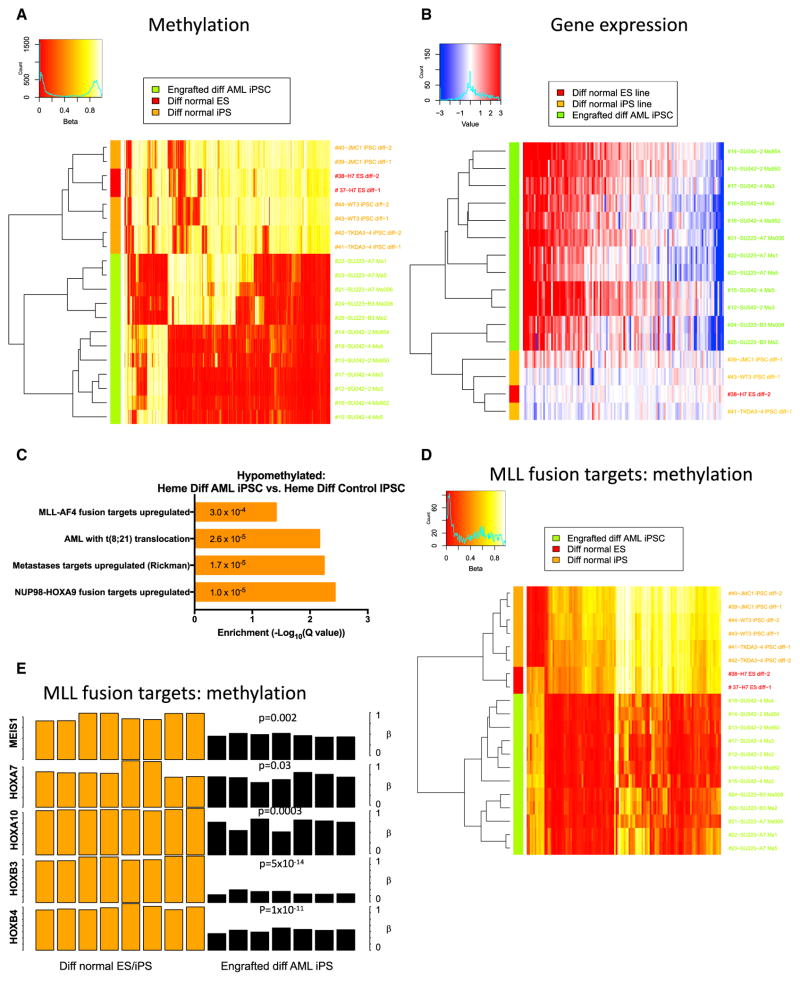
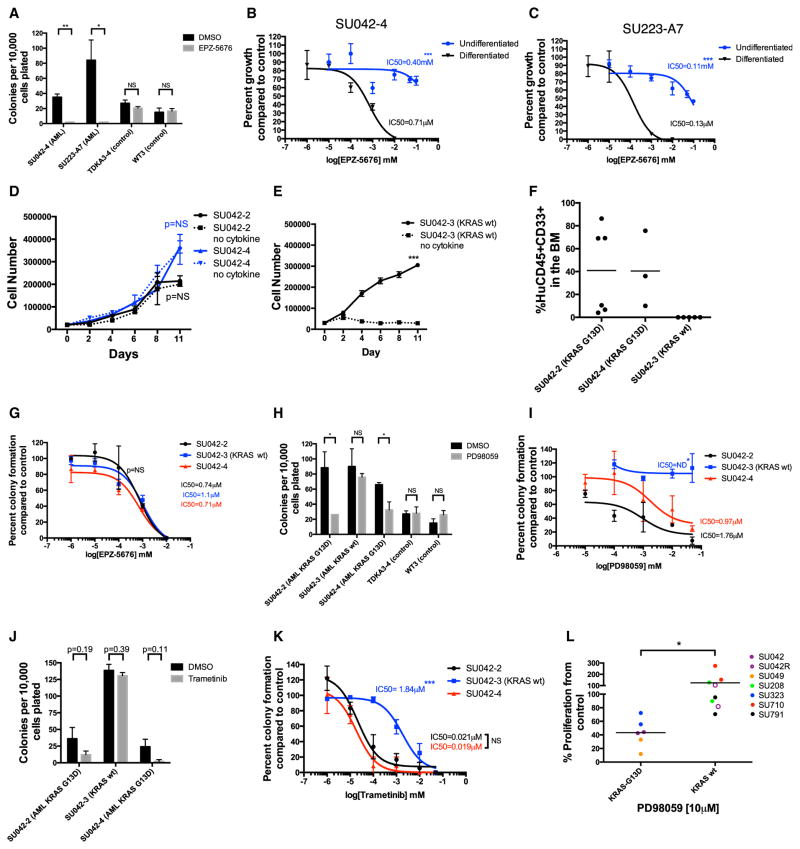
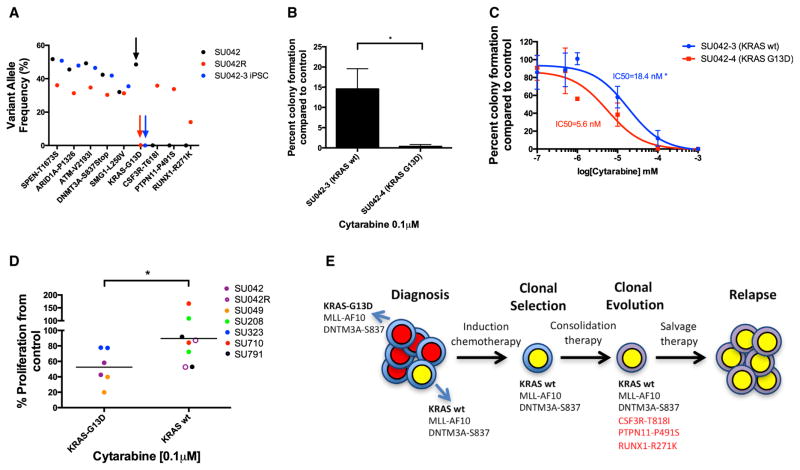
Understanding the relative contributions of genetic and epigenetic abnormalities to acute myeloid leukemia (AML) should assist integrated design of targeted therapies. In this study, we generated induced pluripotent stem cells (iPSCs) from AML patient samples harboring MLL rearrangements and found that they retained leukemic mutations but reset leukemic DNA methylation/gene expression patterns. AML-iPSCs lacked leukemic potential, but when differentiated into hematopoietic cells, they reacquired the ability to give rise to leukemia in vivo and reestablished leukemic DNA methylation/gene expression patterns, including an aberrant MLL signature. Epigenetic reprogramming was therefore not sufficient to eliminate leukemic behavior. This approach also allowed us to study the properties of distinct AML subclones, including differential drug susceptibilities of KRAS mutant and wild-type cells, and predict relapse based on increased cytarabine resistance of a KRAS wild-type subclone. Overall, our findings illustrate the value of AML-iPSCs for investigating the mechanistic basis and clonal properties of human AML.
Keywords: MLL; acute myeloid leukemia; epigenetics; induced pluripotent stem cells; reprogramming.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
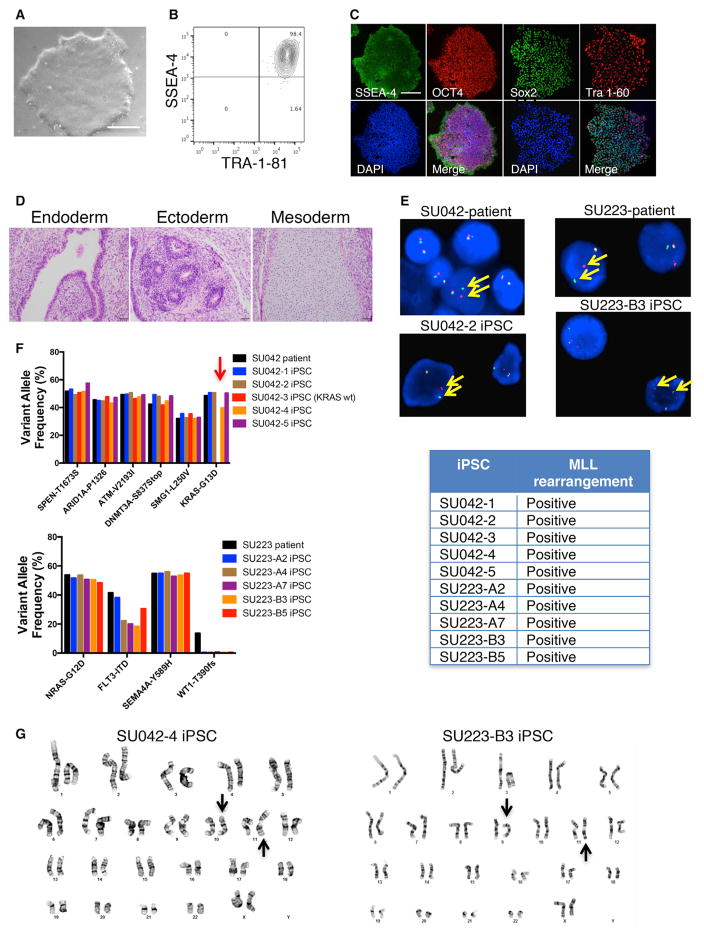
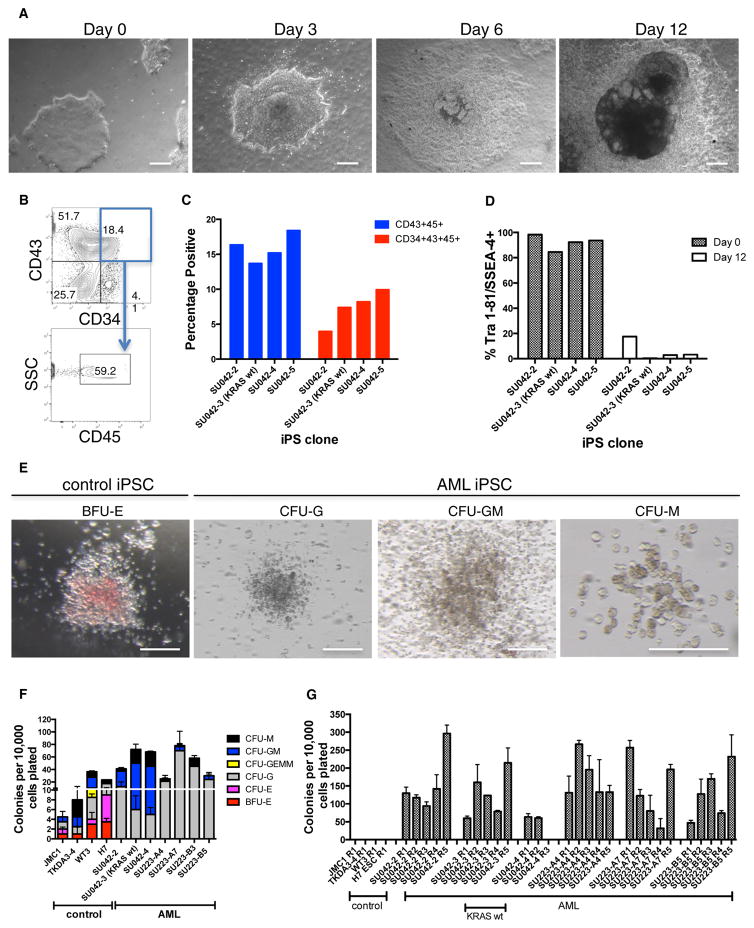
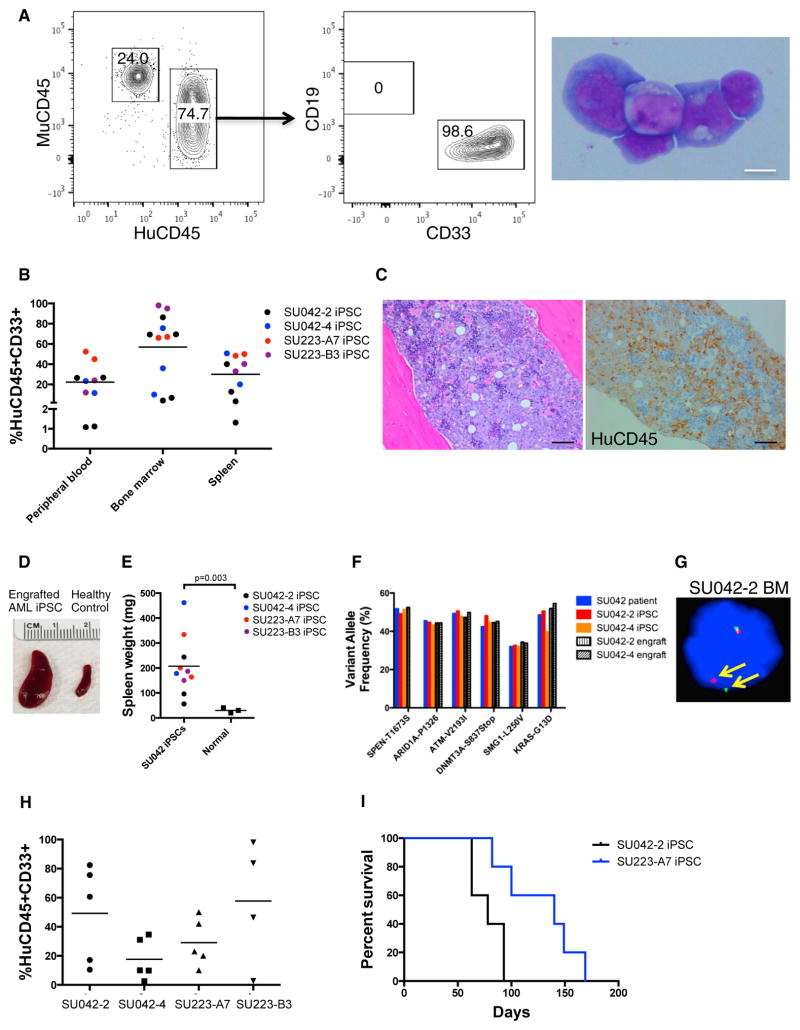
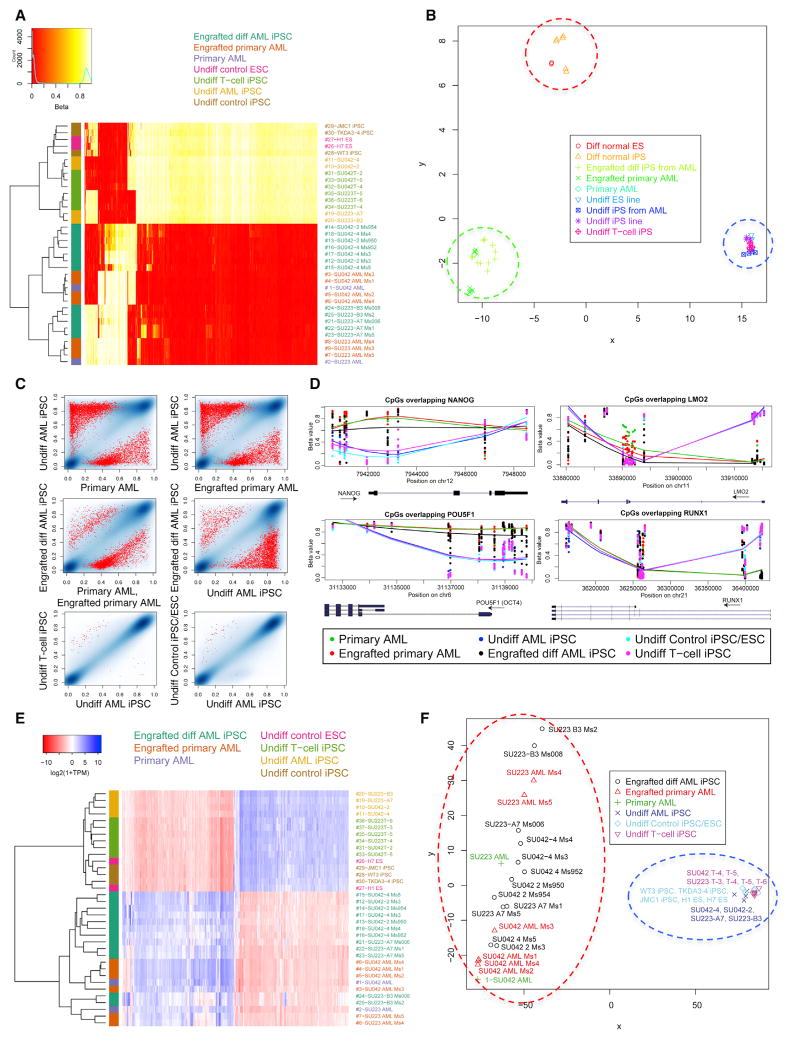
Comment in
-
Bridging the Gaps: iPSC-Based Models from CHIP to MDS to AML.Cell Stem Cell. 2017 Mar 2;20(3):298-299. doi: 10.1016/j.stem.2017.02.011. Cell Stem Cell. 2017. PMID: 28257709
-
Human acute leukemia induced pluripotent stem cells: a unique model for investigating disease development and pathogenesis.Stem Cell Investig. 2017 Jun 13;4:55. doi: 10.21037/sci.2017.05.12. eCollection 2017. Stem Cell Investig. 2017. PMID: 28725651 Free PMC article. No abstract available.
Similar articles
-
Cellular Reprogramming Allows Generation of Autologous Hematopoietic Progenitors From AML Patients That Are Devoid of Patient-Specific Genomic Aberrations.Stem Cells. 2015 Jun;33(6):1839-49. doi: 10.1002/stem.1994. Stem Cells. 2015. PMID: 25764124 Free PMC article.
-
Brief Report: Human Acute Myeloid Leukemia Reprogramming to Pluripotency Is a Rare Event and Selects for Patient Hematopoietic Cells Devoid of Leukemic Mutations.Stem Cells. 2017 Sep;35(9):2095-2102. doi: 10.1002/stem.2655. Epub 2017 Jul 31. Stem Cells. 2017. PMID: 28758276
-
Development Refractoriness of MLL-Rearranged Human B Cell Acute Leukemias to Reprogramming into Pluripotency.Stem Cell Reports. 2016 Oct 11;7(4):602-618. doi: 10.1016/j.stemcr.2016.08.013. Epub 2016 Sep 22. Stem Cell Reports. 2016. PMID: 27666791 Free PMC article.
-
The increasing genomic complexity of acute myeloid leukemia.Best Pract Res Clin Haematol. 2014 Sep-Dec;27(3-4):209-13. doi: 10.1016/j.beha.2014.10.001. Epub 2014 Oct 15. Best Pract Res Clin Haematol. 2014. PMID: 25455268 Review.
-
Epigenetic Guardian: A Review of the DNA Methyltransferase DNMT3A in Acute Myeloid Leukaemia and Clonal Haematopoiesis.Biomed Res Int. 2017;2017:5473197. doi: 10.1155/2017/5473197. Epub 2017 Feb 14. Biomed Res Int. 2017. PMID: 28286768 Free PMC article. Review.
Cited by
-
Modeling Hematological Diseases and Cancer With Patient-Specific Induced Pluripotent Stem Cells.Front Immunol. 2018 Sep 28;9:2243. doi: 10.3389/fimmu.2018.02243. eCollection 2018. Front Immunol. 2018. PMID: 30323816 Free PMC article. Review.
-
Constructing and Deconstructing Cancers using Human Pluripotent Stem Cells and Organoids.Cell Stem Cell. 2019 Jan 3;24(1):12-24. doi: 10.1016/j.stem.2018.11.012. Epub 2018 Dec 20. Cell Stem Cell. 2019. PMID: 30581078 Free PMC article. Review.
-
Modeling blood diseases with human induced pluripotent stem cells.Dis Model Mech. 2019 Jun 4;12(6):dmm039321. doi: 10.1242/dmm.039321. Dis Model Mech. 2019. PMID: 31171568 Free PMC article. Review.
-
Murine Models of Acute Myeloid Leukaemia.Int J Mol Sci. 2019 Jan 21;20(2):453. doi: 10.3390/ijms20020453. Int J Mol Sci. 2019. PMID: 30669675 Free PMC article. Review.
-
Engineering considerations of iPSC-based personalized medicine.Biomater Res. 2023 Jul 7;27(1):67. doi: 10.1186/s40824-023-00382-x. Biomater Res. 2023. PMID: 37420273 Free PMC article. Review.
References
-
- Afonja O, Smith JE, Jr, Cheng DM, Goldenberg AS, Amorosi E, Shimamoto T, Nakamura S, Ohyashiki K, Ohyashiki J, Toyama K, Takeshita K. MEIS1 and HOXA7 genes in human acute myeloid leukemia. Leuk Res. 2000;24:849–855. - PubMed
-
- Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27:1000–1008. - PubMed
-
- Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–6776. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
