

Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway
- PMID: 28060376
- PMCID: PMC5384020
- DOI: 10.1038/cdd.2016.153
Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway
Abstract
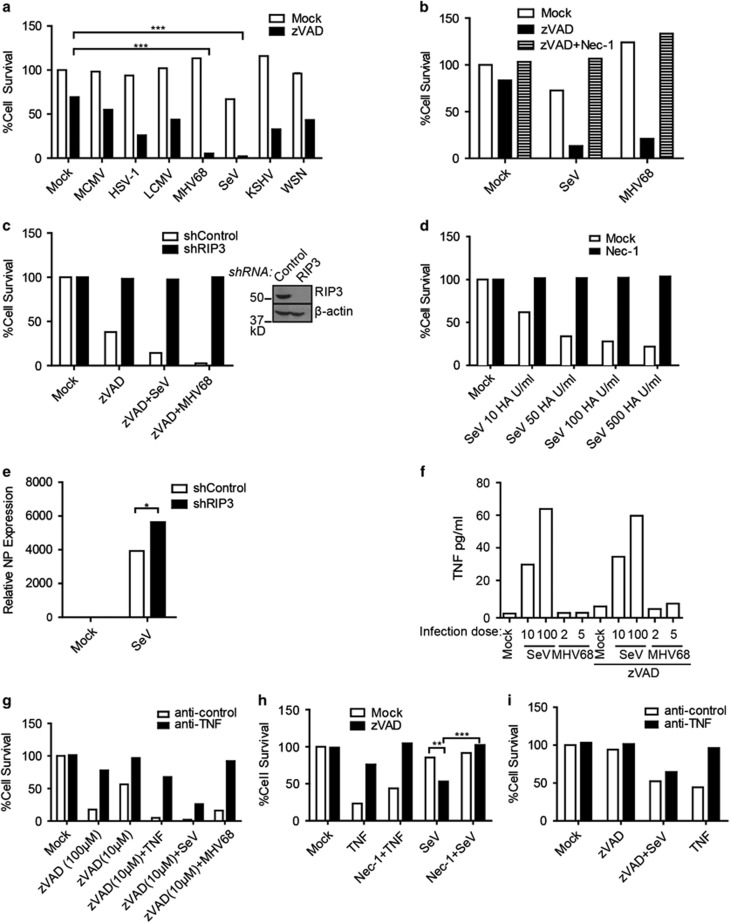
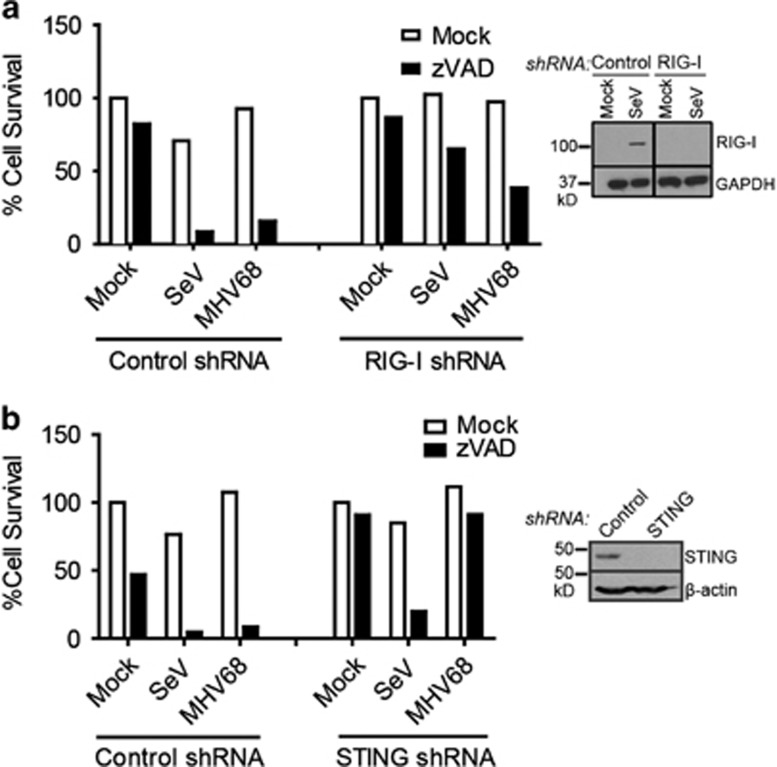
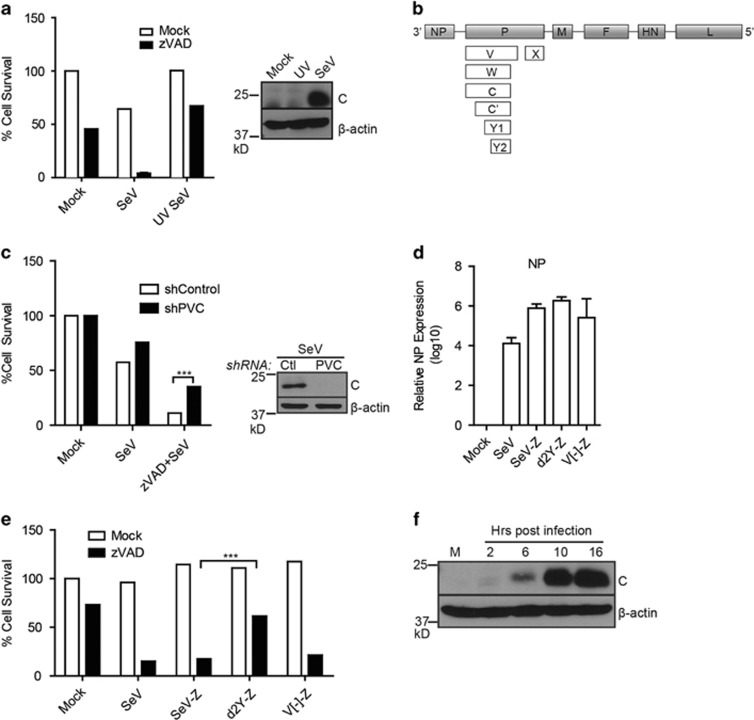
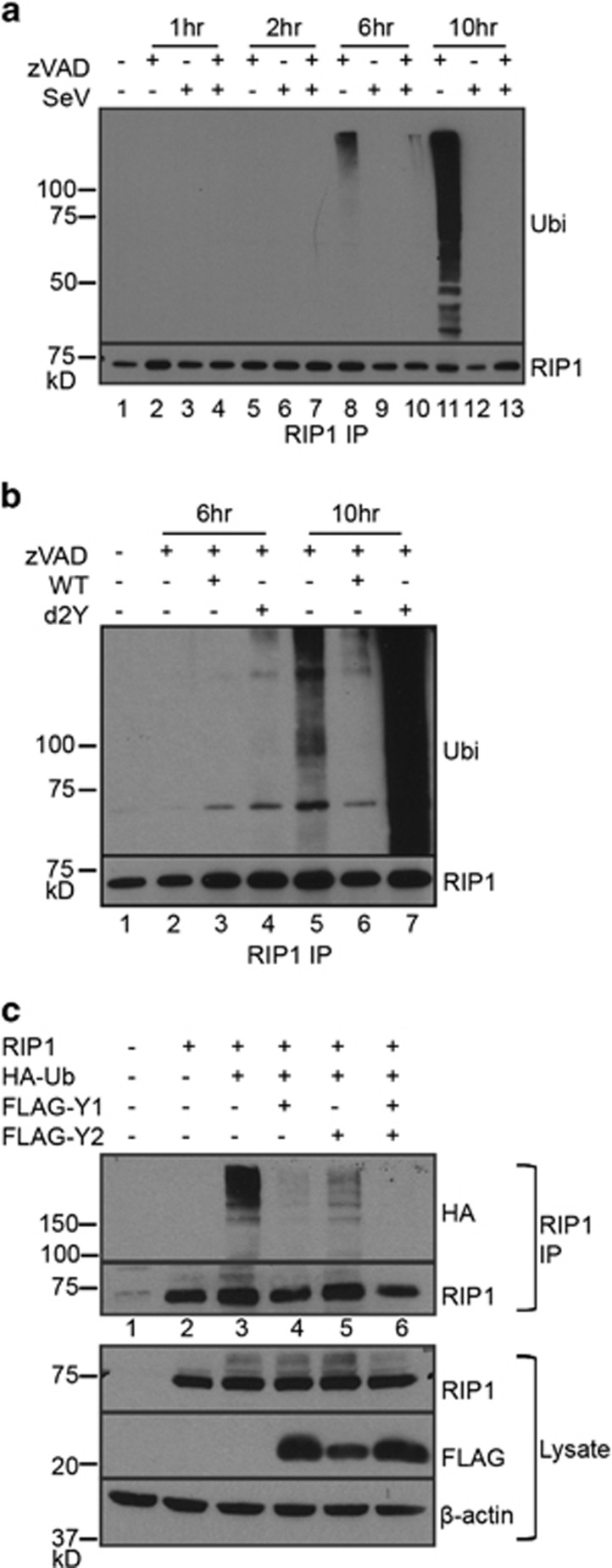
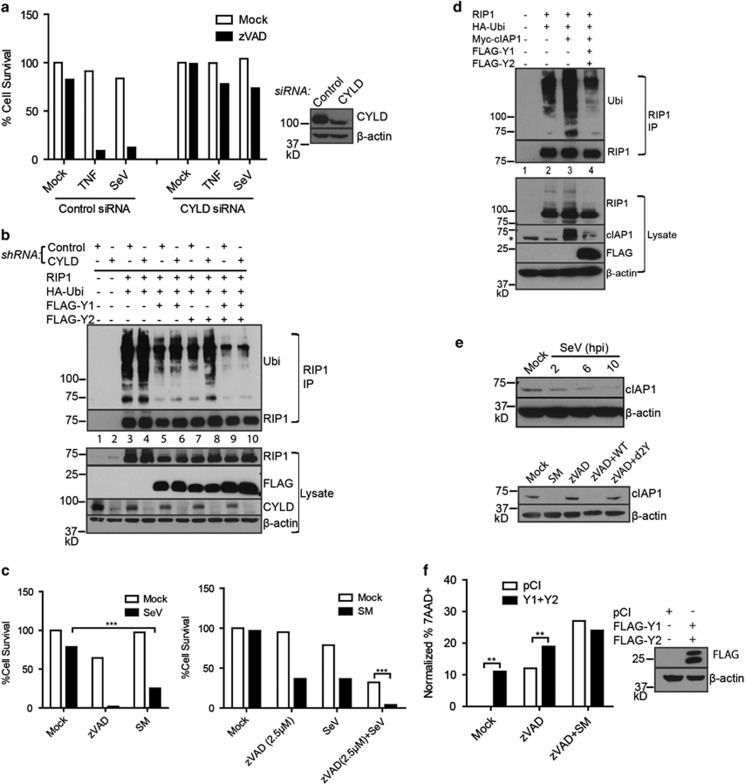
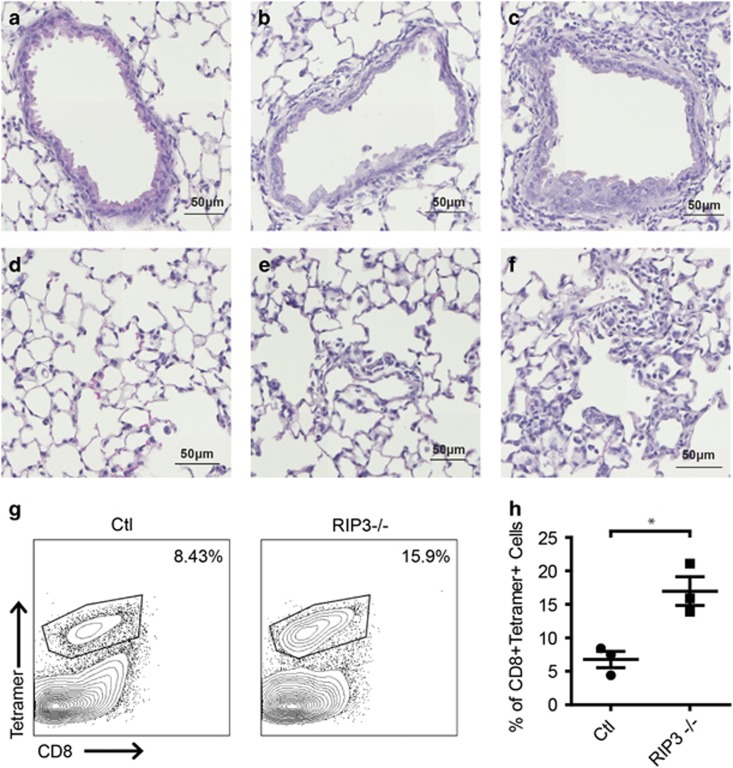
Necroptosis is a form of necrotic cell death that requires the activity of the death domain-containing kinase RIP1 and its family member RIP3. Necroptosis occurs when RIP1 is deubiquitinated to form a complex with RIP3 in cells deficient in the death receptor adapter molecule FADD or caspase-8. Necroptosis may play a role in host defense during viral infection as viruses like vaccinia can induce necroptosis while murine cytomegalovirus encodes a viral inhibitor of necroptosis. To see how general the interplay between viruses and necroptosis is, we surveyed seven different viruses. We found that two of the viruses tested, Sendai virus (SeV) and murine gammaherpesvirus-68 (MHV68), are capable of inducing dramatic necroptosis in the fibrosarcoma L929 cell line. We show that MHV68-induced cell death occurs through the cytosolic STING sensor pathway in a TNF-dependent manner. In contrast, SeV-induced death is mostly independent of TNF. Knockdown of the RNA sensing molecule RIG-I or the RIP1 deubiquitin protein, CYLD, but not STING, rescued cells from SeV-induced necroptosis. Accompanying necroptosis, we also find that wild type but not mutant SeV lacking the viral proteins Y1 and Y2 result in the non-ubiquitinated form of RIP1. Expression of Y1 or Y2 alone can suppress RIP1 ubiquitination but CYLD is dispensable for this process. Instead, we found that Y1 and Y2 can inhibit cIAP1-mediated RIP1 ubiquitination. Interestingly, we also found that SeV infection of B6 RIP3-/- mice results in increased inflammation in the lung and elevated SeV-specific T cells. Collectively, these data identify viruses and pathways that can trigger necroptosis and highlight the dynamic interplay between pathogen-recognition receptors and cell death induction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis.PLoS One. 2013 Oct 2;8(10):e76841. doi: 10.1371/journal.pone.0076841. eCollection 2013. PLoS One. 2013. PMID: 24098568 Free PMC article.
-
cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production.Cell Death Differ. 2011 Apr;18(4):656-65. doi: 10.1038/cdd.2010.138. Epub 2010 Nov 5. Cell Death Differ. 2011. PMID: 21052097 Free PMC article.
-
Cylindromatosis mediates neuronal cell death in vitro and in vivo.Cell Death Differ. 2018 Aug;25(8):1394-1407. doi: 10.1038/s41418-017-0046-7. Epub 2018 Jan 19. Cell Death Differ. 2018. PMID: 29352268 Free PMC article.
-
Necroptosis in health and diseases.Semin Cell Dev Biol. 2014 Nov;35:14-23. doi: 10.1016/j.semcdb.2014.07.013. Epub 2014 Aug 1. Semin Cell Dev Biol. 2014. PMID: 25087983 Review.
-
RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review).Int J Mol Med. 2019 Sep;44(3):771-786. doi: 10.3892/ijmm.2019.4244. Epub 2019 Jun 14. Int J Mol Med. 2019. PMID: 31198981 Free PMC article. Review.
Cited by
-
TNFα promotes glioblastoma A172 cell mitochondrial apoptosis via augmenting mitochondrial fission and repression of MAPK-ERK-YAP signaling pathways.Onco Targets Ther. 2018 Oct 18;11:7213-7227. doi: 10.2147/OTT.S184337. eCollection 2018. Onco Targets Ther. 2018. Retraction in: Onco Targets Ther. 2024 Apr 03;17:297-298. doi: 10.2147/OTT.S471180. PMID: 30425514 Free PMC article. Retracted.
-
Ripk3 signaling regulates HSCs during stress and represses radiation-induced leukemia in mice.Stem Cell Reports. 2022 Jun 14;17(6):1428-1441. doi: 10.1016/j.stemcr.2022.04.009. Epub 2022 May 12. Stem Cell Reports. 2022. PMID: 35561683 Free PMC article.
-
Inhibition of murine herpesvirus-68 replication by IFN-gamma in macrophages is counteracted by the induction of SOCS1 expression.PLoS Pathog. 2018 Aug 3;14(8):e1007202. doi: 10.1371/journal.ppat.1007202. eCollection 2018 Aug. PLoS Pathog. 2018. PMID: 30075008 Free PMC article.
-
Innate immune response restarts adaptive immune response in tumors.Front Immunol. 2023 Sep 14;14:1260705. doi: 10.3389/fimmu.2023.1260705. eCollection 2023. Front Immunol. 2023. PMID: 37781382 Free PMC article. Review.
-
Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines.Cancer Cell. 2018 Apr 9;33(4):599-605. doi: 10.1016/j.ccell.2018.03.011. Cancer Cell. 2018. PMID: 29634947 Free PMC article. Review.
References
-
- Blander JM. A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat Rev Immunol 2014; 14: 601–618. - PubMed
-
- Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: toward identification of the effector molecules. Science 2016; 352: aaf2154. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
