

Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance
- PMID: 27989769
- PMCID: PMC5291795
- DOI: 10.1016/j.stem.2016.11.003
Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance
Abstract
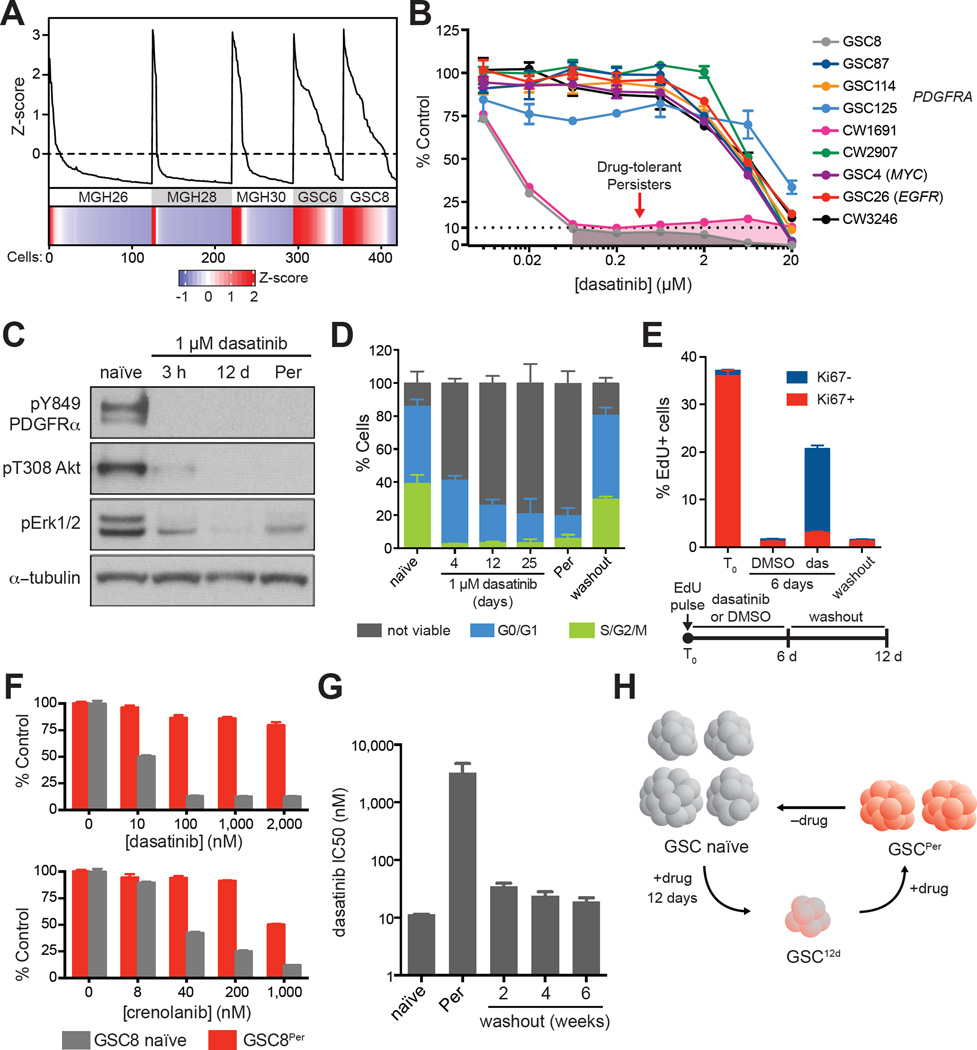
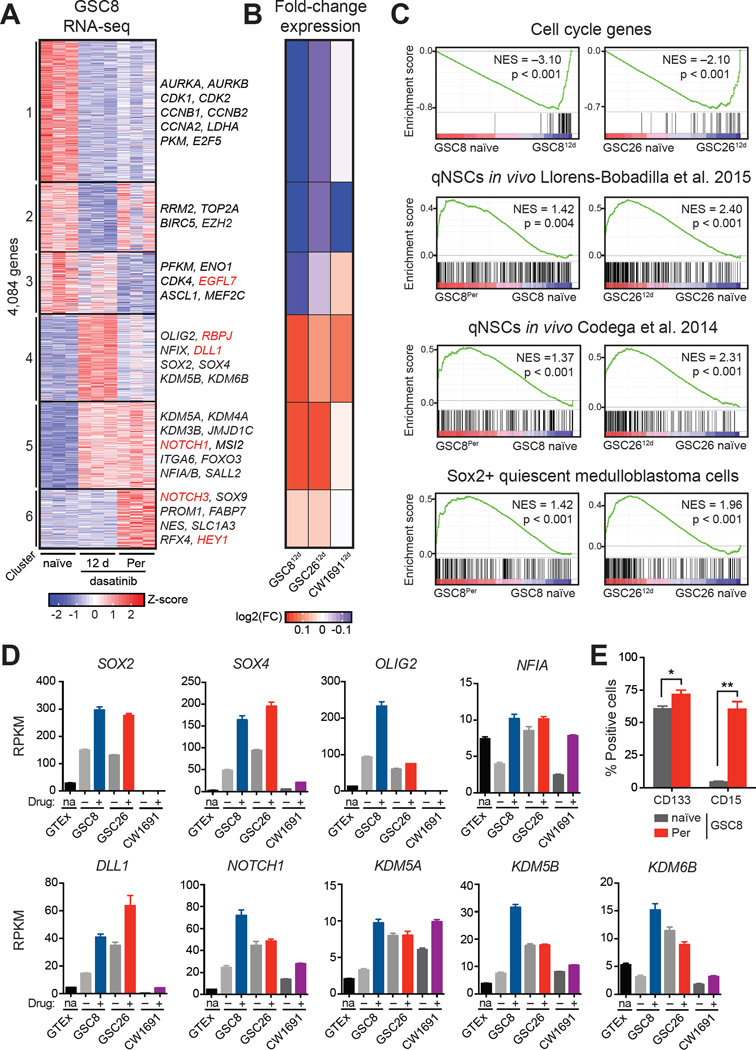
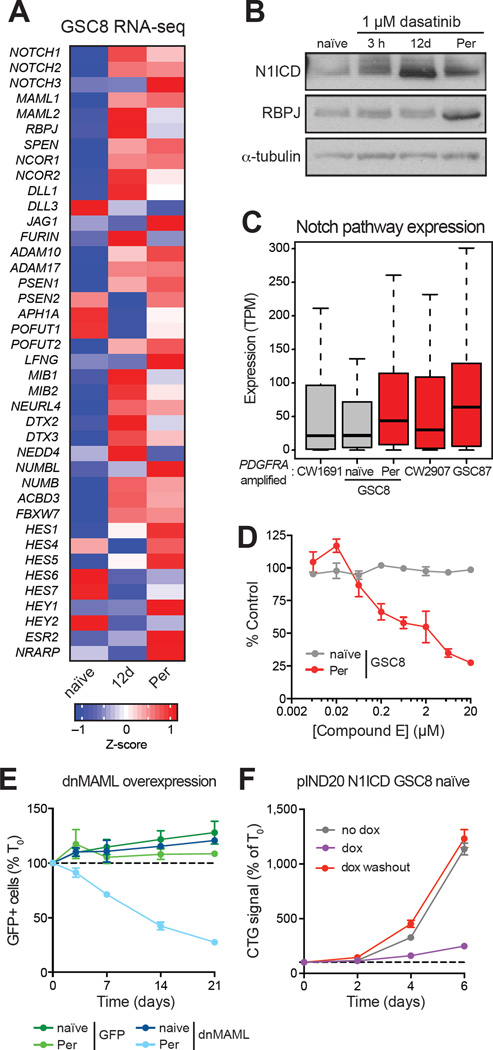
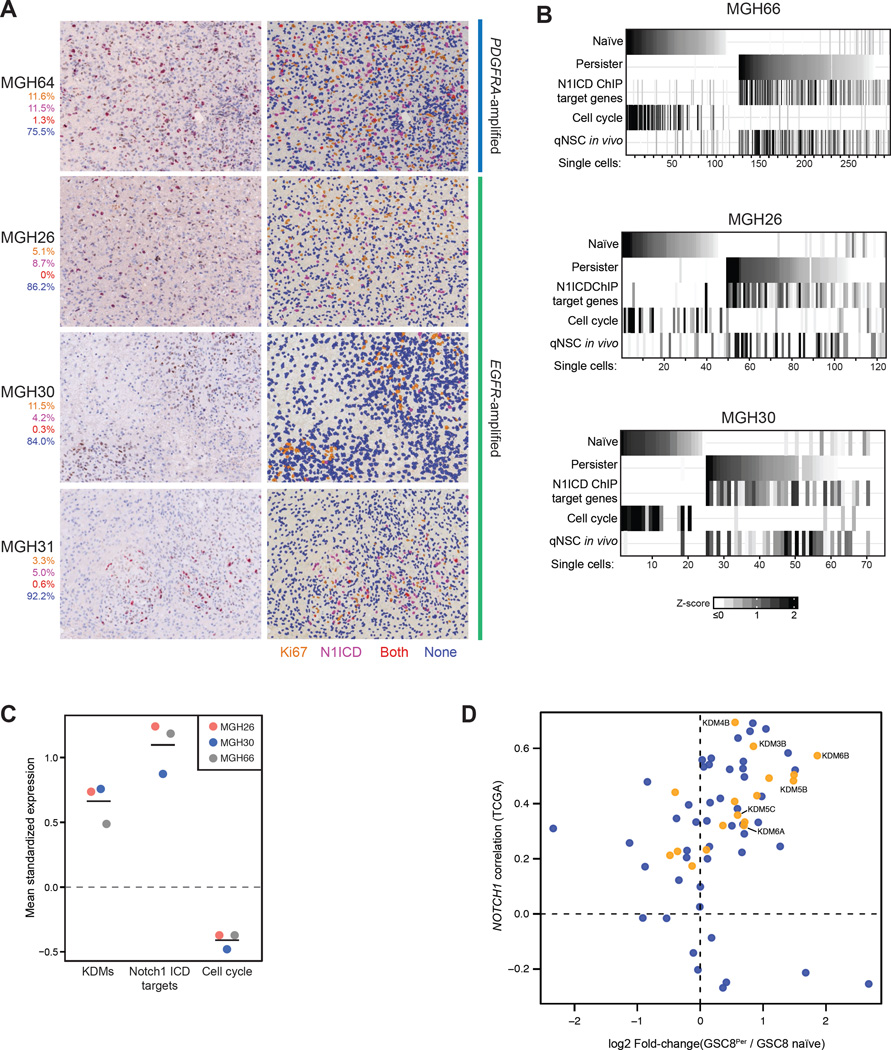
Glioblastoma, the most common and aggressive malignant brain tumor, is propagated by stem-like cancer cells refractory to existing therapies. Understanding the molecular mechanisms that control glioblastoma stem cell (GSC) proliferation and drug resistance may reveal opportunities for therapeutic interventions. Here we show that GSCs can reversibly transition to a slow-cycling, persistent state in response to targeted kinase inhibitors. In this state, GSCs upregulate primitive developmental programs and are dependent upon Notch signaling. This transition is accompanied by widespread redistribution of repressive histone methylation. Accordingly, persister GSCs upregulate, and are dependent on, the histone demethylases KDM6A/B. Slow-cycling cells with high Notch activity and histone demethylase expression are present in primary glioblastomas before treatment, potentially contributing to relapse. Our findings illustrate how cancer cells may hijack aspects of native developmental programs for deranged proliferation, adaptation, and tolerance. They also suggest strategies for eliminating refractory tumor cells by targeting epigenetic and developmental pathways.
Keywords: KDM6A/B; Notch; cancer; chromatin; drug resistance; epigenetics; glioblastoma; histone demethylases; stem cell.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B.Mol Oncol. 2018 Mar;12(3):406-420. doi: 10.1002/1878-0261.12174. Epub 2018 Feb 12. Mol Oncol. 2018. PMID: 29360266 Free PMC article.
-
The role of histone H3 lysine demethylases in glioblastoma.Cancer Metastasis Rev. 2023 Jun;42(2):445-454. doi: 10.1007/s10555-023-10114-1. Epub 2023 Jun 8. Cancer Metastasis Rev. 2023. PMID: 37286866 Review.
-
Glioblastoma initiating cells are sensitive to histone demethylase inhibition due to epigenetic deregulation.Int J Cancer. 2020 Mar 1;146(5):1281-1292. doi: 10.1002/ijc.32649. Epub 2019 Oct 11. Int J Cancer. 2020. PMID: 31456217
-
Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo.Cell Death Dis. 2014 May 8;5(5):e1223. doi: 10.1038/cddis.2014.188. Cell Death Dis. 2014. PMID: 24810059 Free PMC article.
-
Histone demethylase lysine demethylase 5B in development and cancer.Oncotarget. 2017 Jan 31;8(5):8980-8991. doi: 10.18632/oncotarget.13858. Oncotarget. 2017. PMID: 27974677 Free PMC article. Review.
Cited by
-
De novo purine biosynthesis is a major driver of chemoresistance in glioblastoma.Brain. 2021 May 7;144(4):1230-1246. doi: 10.1093/brain/awab020. Brain. 2021. PMID: 33855339 Free PMC article.
-
Targeted inhibition of KDM6 histone demethylases eradicates tumor-initiating cells via enhancer reprogramming in colorectal cancer.Theranostics. 2020 Aug 8;10(22):10016-10030. doi: 10.7150/thno.47081. eCollection 2020. Theranostics. 2020. PMID: 32929331 Free PMC article.
-
Liquid biopsy-based single-cell metabolic phenotyping of lung cancer patients for informative diagnostics.Nat Commun. 2019 Aug 26;10(1):3856. doi: 10.1038/s41467-019-11808-3. Nat Commun. 2019. PMID: 31451693 Free PMC article.
-
Acquired Resistance to HER2-Targeted Therapies Creates Vulnerability to ATP Synthase Inhibition.Cancer Res. 2020 Feb 1;80(3):524-535. doi: 10.1158/0008-5472.CAN-18-3985. Epub 2019 Nov 5. Cancer Res. 2020. PMID: 31690671 Free PMC article.
-
Cancer stem cells in brain tumors: From origin to clinical implications.MedComm (2020). 2023 Aug 9;4(4):e341. doi: 10.1002/mco2.341. eCollection 2023 Aug. MedComm (2020). 2023. PMID: 37576862 Free PMC article. Review.
References
-
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. - PubMed
-
- Black BL, Ligon KL, Zhang Y, Olson EN. Cooperative transcriptional activation by the neurogenic basic helix-loop-helix protein MASH1 and members of the myocyte enhancer factor-2 (MEF2) family. Journal of Biological Chemistry. 1996;271:26659–26663. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
