

Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells
- PMID: 27983869
- PMCID: PMC5449492
- DOI: 10.1165/rcmb.2016-0276MA
Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells
Abstract
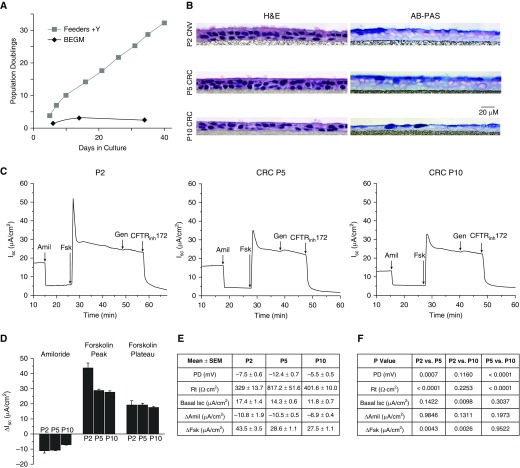
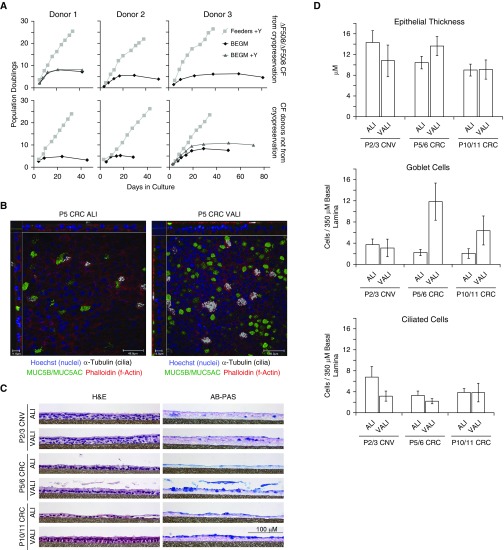
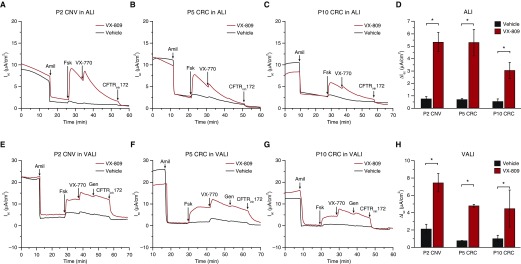
Well-differentiated primary human bronchial epithelial (HBE) cell cultures are vital for cystic fibrosis (CF) research, particularly for the development of cystic fibrosis transmembrane conductance regulator (CFTR) modulator drugs. Culturing of epithelial cells with irradiated 3T3 fibroblast feeder cells plus the RhoA kinase inhibitor Y-27632 (Y), termed conditionally reprogrammed cell (CRC) technology, enhances cell growth and lifespan while preserving cell-of-origin functionality. We initially determined the electrophysiological and morphological characteristics of conventional versus CRC-expanded non-CF HBE cells. On the basis of these findings, we then created six CF cell CRC populations, three from sequentially obtained CF lungs and three from F508 del homozygous donors previously obtained and cryopreserved using conventional culture methods. Growth curves were plotted, and cells were subcultured, without irradiated feeders plus Y, into air-liquid interface conditions in nonproprietary and proprietary Ultroser G-containing media and were allowed to differentiate. Ussing chamber studies were performed after treatment of F508 del homozygous CF cells with the CFTR modulator VX-809. Bronchial epithelial cells grew exponentially in feeders plus Y, dramatically surpassing the numbers of conventionally grown cells. Passage 5 and 10 CRC HBE cells formed confluent mucociliary air-liquid interface cultures. There were differences in cell morphology and current magnitude as a function of extended passage, but the effect of VX-809 in increasing CFTR function was significant in CRC-expanded F508 del HBE cells. Thus, CRC technology expands the supply of functional primary CF HBE cells for testing CFTR modulators in Ussing chambers.
Keywords: cystic fibrosis; electrophysiology; human bronchial epithelial cells; in vitro models.
Figures
Similar articles
-
Assessing Human Airway Epithelial Progenitor Cells for Cystic Fibrosis Cell Therapy.Am J Respir Cell Mol Biol. 2020 Sep;63(3):374-385. doi: 10.1165/rcmb.2019-0384OC. Am J Respir Cell Mol Biol. 2020. PMID: 32437238 Free PMC article.
-
In vitro 3D culture lung model from expanded primary cystic fibrosis human airway cells.J Cyst Fibros. 2020 Sep;19(5):752-761. doi: 10.1016/j.jcf.2020.05.007. Epub 2020 Jun 18. J Cyst Fibros. 2020. PMID: 32565193 Free PMC article.
-
Significant functional differences in differentiated Conditionally Reprogrammed (CRC)- and Feeder-free Dual SMAD inhibited-expanded human nasal epithelial cells.J Cyst Fibros. 2021 Mar;20(2):364-371. doi: 10.1016/j.jcf.2020.12.019. Epub 2021 Jan 5. J Cyst Fibros. 2021. PMID: 33414087
-
Long-term differentiating primary human airway epithelial cell cultures: how far are we?Cell Commun Signal. 2021 May 27;19(1):63. doi: 10.1186/s12964-021-00740-z. Cell Commun Signal. 2021. PMID: 34044844 Free PMC article. Review.
-
Modeling respiratory tract diseases for clinical translation employing conditionally reprogrammed cells.Cell Insight. 2024 Sep 18;3(6):100201. doi: 10.1016/j.cellin.2024.100201. eCollection 2024 Dec. Cell Insight. 2024. PMID: 39391007 Free PMC article. Review.
Cited by
-
Airway Epithelial Inflammation In Vitro Augments the Rescue of Mutant CFTR by Current CFTR Modulator Therapies.Front Pharmacol. 2021 Mar 30;12:628722. doi: 10.3389/fphar.2021.628722. eCollection 2021. Front Pharmacol. 2021. PMID: 33859562 Free PMC article.
-
Bioengineering Human Upper Respiratory Mucosa: A Systematic Review of the State of the Art of Cell Culture Techniques.Bioengineering (Basel). 2024 Aug 13;11(8):826. doi: 10.3390/bioengineering11080826. Bioengineering (Basel). 2024. PMID: 39199784 Free PMC article. Review.
-
Assessing Human Airway Epithelial Progenitor Cells for Cystic Fibrosis Cell Therapy.Am J Respir Cell Mol Biol. 2020 Sep;63(3):374-385. doi: 10.1165/rcmb.2019-0384OC. Am J Respir Cell Mol Biol. 2020. PMID: 32437238 Free PMC article.
-
SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract.Cell. 2020 Jul 23;182(2):429-446.e14. doi: 10.1016/j.cell.2020.05.042. Epub 2020 May 27. Cell. 2020. PMID: 32526206 Free PMC article.
-
CFTR bearing variant p.Phe312del exhibits function inconsistent with phenotype and negligible response to ivacaftor.JCI Insight. 2022 Mar 22;7(6):e148841. doi: 10.1172/jci.insight.148841. JCI Insight. 2022. PMID: 35315358 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
