

Kidney Fibrosis: Origins and Interventions
- PMID: 27941433
- PMCID: PMC7228593
- DOI: 10.1097/TP.0000000000001608
Kidney Fibrosis: Origins and Interventions
Abstract
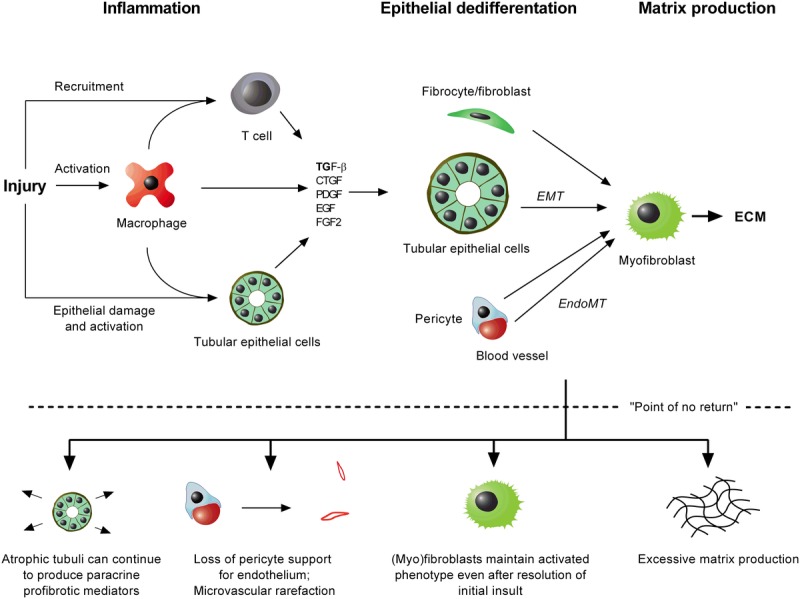
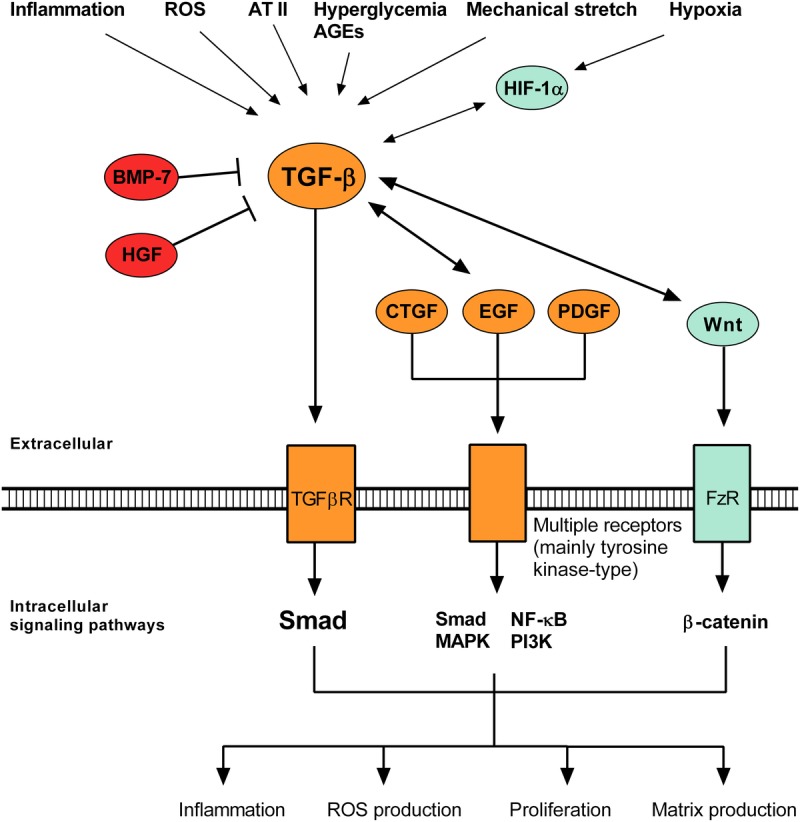
All causes of renal allograft injury, when severe and/or sustained, can result in chronic histological damage of which interstitial fibrosis and tubular atrophy are dominant features. Unless a specific disease process can be identified, what drives interstitial fibrosis and tubular atrophy progression in individual patients is often unclear. In general, clinicopathological factors known to predict and drive allograft fibrosis include graft quality, inflammation (whether "nonspecific" or related to a specific diagnosis), infections, such as polyomavirus-associated nephropathy, calcineurin inhibitors (CNI), and genetic factors. The incidence and severity of chronic histological damage have decreased substantially over the last 3 decades, but it is difficult to disentangle what effects individual innovations (eg, better matching and preservation techniques, lower CNI dosing, BK viremia screening) may have had. There is little evidence that CNI-sparing/minimization strategies, steroid minimization or renin-angiotensin-aldosterone system blockade result in better preservation of intermediate-term histology. Treatment of subclinical rejections has only proven beneficial to histological and functional outcome in studies in which the rate of subclinical rejection in the first 3 months was greater than 10% to 15%. Potential novel antifibrotic strategies include antagonists of transforming growth factor-β, connective tissue growth factor, several tyrosine kinase ligands (epidermal growth factor, platelet-derived growth factor, vascular endothelial growth factor), endothelin and inhibitors of chemotaxis. Although many of these drugs are mainly being developed and marketed for oncological indications and diseases, such as idiopathic pulmonary fibrosis, a number may hold promise in the treatment of diabetic nephropathy, which could eventually lead to applications in renal transplantation.
Conflict of interest statement
The authors declare no funding or conflicts of interest.
Figures
Similar articles
-
Renin-Angiotensin System Blockage and Avoiding High Doses of Calcineurin Inhibitors Prevent Interstitial Fibrosis and Tubular Atrophy in Kidney Transplant Recipients.Exp Clin Transplant. 2017 Feb;15(Suppl 1):32-36. doi: 10.6002/ect.mesot2016.O19. Exp Clin Transplant. 2017. PMID: 28260428
-
Early withdrawal of calcineurin inhibitor from a sirolimus-based immunosuppression stabilizes fibrosis and the transforming growth factor-β signalling pathway in kidney transplant.Nephrology (Carlton). 2015 Mar;20(3):168-76. doi: 10.1111/nep.12368. Nephrology (Carlton). 2015. PMID: 25404086 Clinical Trial.
-
Treatment strategies to minimize or prevent chronic allograft dysfunction in pediatric renal transplant recipients: an overview.Paediatr Drugs. 2009;11(6):381-96. doi: 10.2165/11316100-000000000-00000. Paediatr Drugs. 2009. PMID: 19877724 Review.
-
Chronic allograft dysfunction: can we use mammalian target of rapamycin inhibitors to replace calcineurin inhibitors to preserve graft function?Curr Opin Organ Transplant. 2008 Dec;13(6):614-21. doi: 10.1097/MOT.0b013e3283193bad. Curr Opin Organ Transplant. 2008. PMID: 19060552 Review.
-
Chronic Allograft Injury.Clin J Am Soc Nephrol. 2021 Nov;16(11):1723-1729. doi: 10.2215/CJN.15590920. Epub 2021 Apr 5. Clin J Am Soc Nephrol. 2021. PMID: 33820759 Free PMC article. Review.
Cited by
-
Association between post-transplant uric acid level and renal allograft fibrosis: Analysis using Banff pathologic scores from renal biopsies.Sci Rep. 2018 Aug 2;8(1):11601. doi: 10.1038/s41598-018-29948-9. Sci Rep. 2018. PMID: 30072753 Free PMC article.
-
Biomarkers and Pharmacogenomics in Kidney Transplantation.Mol Diagn Ther. 2018 Oct;22(5):537-550. doi: 10.1007/s40291-018-0349-5. Mol Diagn Ther. 2018. PMID: 29971647 Review.
-
Rictor/mTORC2 signalling contributes to renal vascular endothelial-to-mesenchymal transition and renal allograft interstitial fibrosis by regulating BNIP3-mediated mitophagy.Clin Transl Med. 2024 May;14(5):e1686. doi: 10.1002/ctm2.1686. Clin Transl Med. 2024. PMID: 38769658 Free PMC article.
-
The Presence of Urinary Renal Progenitor Cells in Stable Kidney Transplant Recipients Anticipates Allograft Deterioration.Front Physiol. 2018 Oct 10;9:1412. doi: 10.3389/fphys.2018.01412. eCollection 2018. Front Physiol. 2018. PMID: 30364198 Free PMC article.
-
Curcumin Blunts IL-6 Dependent Endothelial-to-Mesenchymal Transition to Alleviate Renal Allograft Fibrosis Through Autophagy Activation.Front Immunol. 2021 May 28;12:656242. doi: 10.3389/fimmu.2021.656242. eCollection 2021. Front Immunol. 2021. Retraction in: Front Immunol. 2021 Nov 26;12:818539. doi: 10.3389/fimmu.2021.818539. PMID: 34122411 Free PMC article. Retracted.
References
-
- Meng XM, Nikolic-paterson DJ, Lan HY. Inflammatory processes in renal fibrosis Nat Rev Nephrol 2014. 10493–503 - PubMed
-
- Boor P, Floege J. Renal allograft fibrosis: biology and therapeutic targets Am J Transplant 2015. 15863–886 - PubMed
-
- Falke LL, Gholizadeh S, Goldschmeding R. Diverse origins of the myofibroblast—implications for kidney fibrosis Nat Rev Nephrol 2015. 11233–244 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
