

Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas
- PMID: 27911794
- PMCID: PMC5167176
- DOI: 10.1073/pnas.1611106113
Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas
Abstract
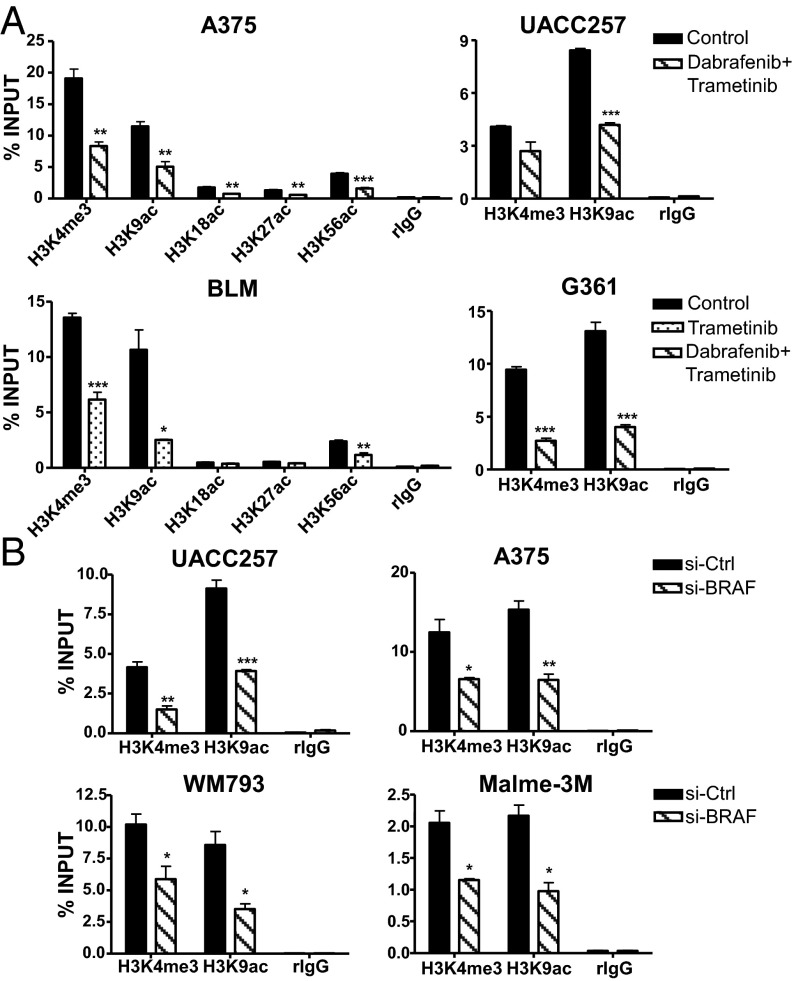
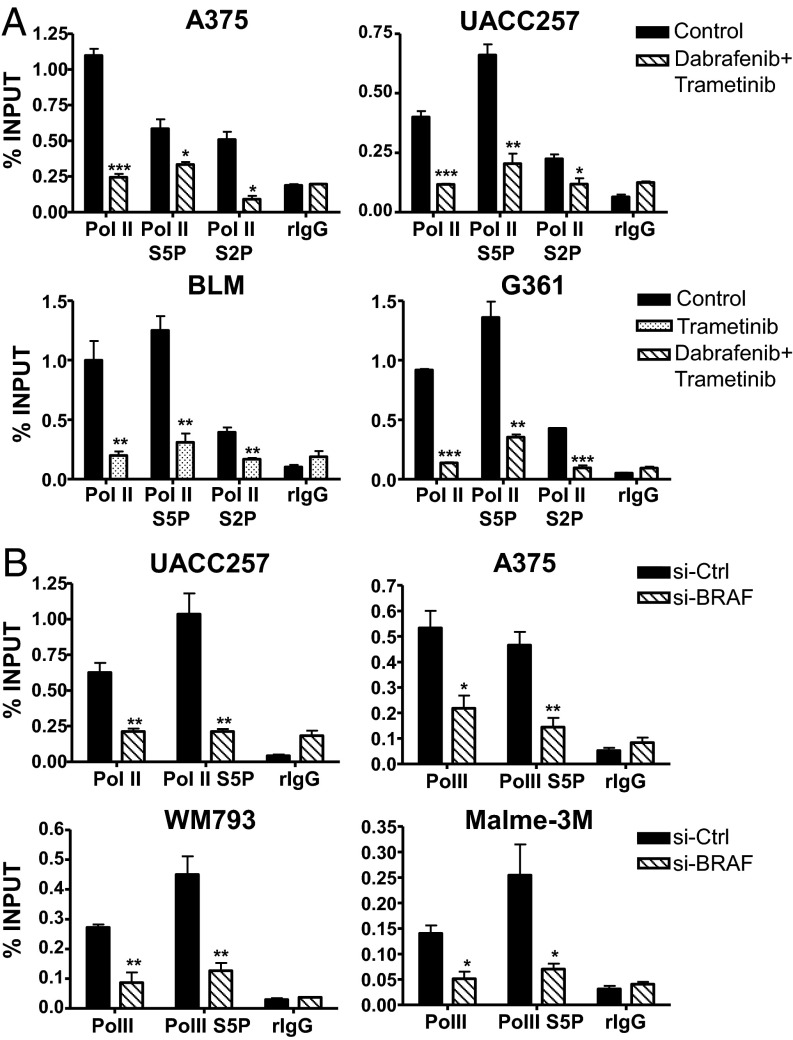
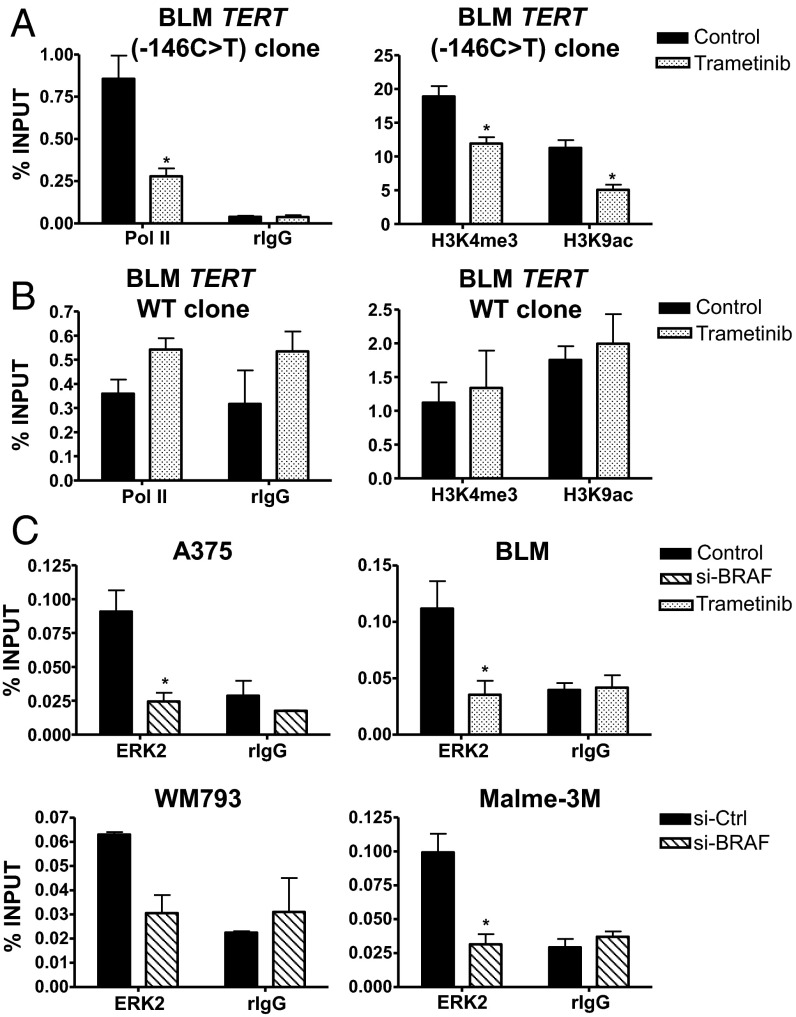
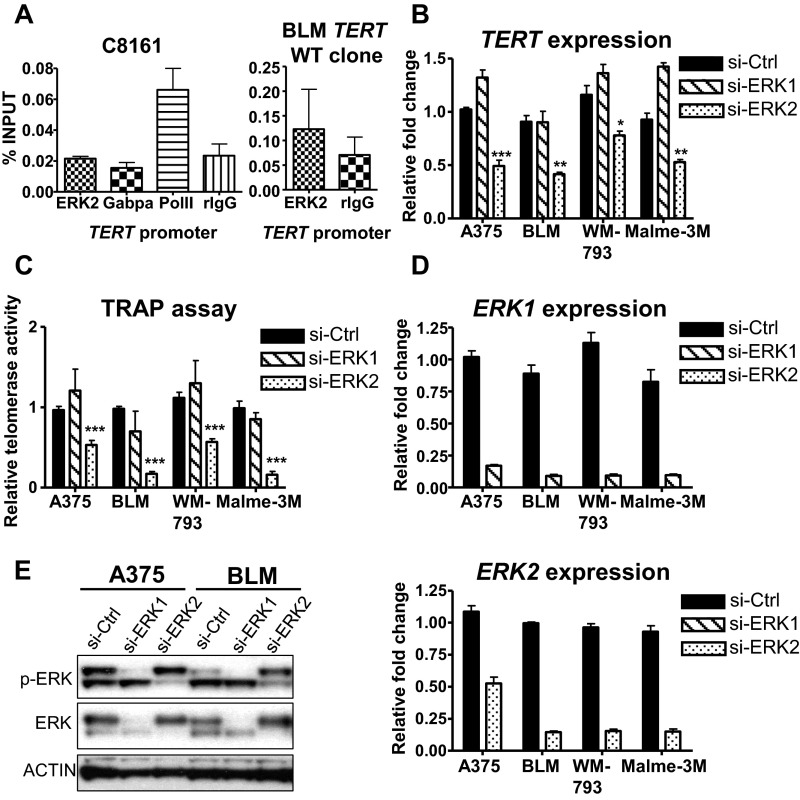
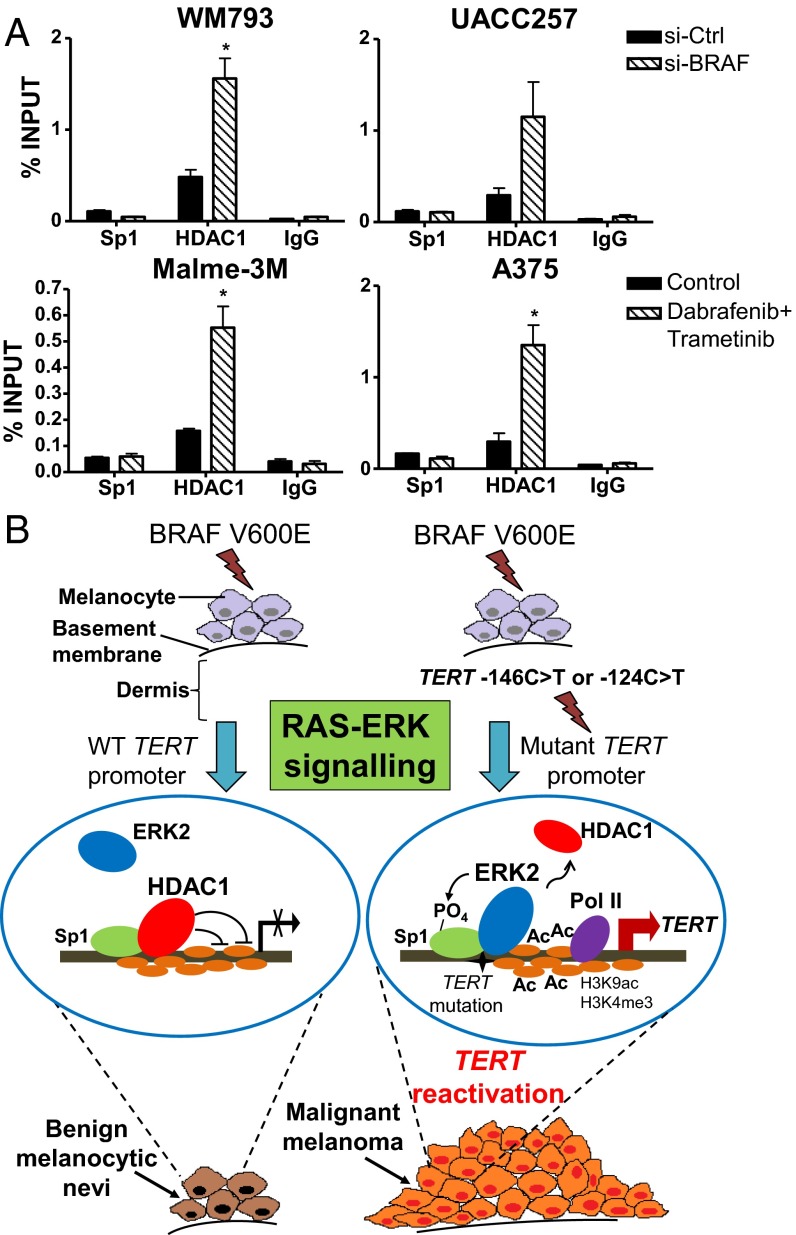
Although activating BRAF/NRAS mutations are frequently seen in melanomas, they are not sufficient to drive malignant transformation and require additional events. Frequent co-occurrence of mutations in the promoter for telomerase reverse transcriptase (TERT), along with BRAF alterations, has recently been noted and correlated with poorer prognosis, implicating a functional link between BRAF signaling and telomerase reactivation in melanomas. Here, we report that RAS-ERK signaling in BRAF mutant melanomas is critical for regulating active chromatin state and recruitment of RNA polymerase II at mutant TERT promoters. Our study provides evidence that the mutant TERT promoter is a key substrate downstream of the RAS-ERK pathway. Reactivating TERT and hence reconstituting telomerase is an important step in melanoma progression from nonmalignant nevi with BRAF mutations. Hence, combined targeting of RAS-ERK and TERT promoter remodeling is a promising avenue to limit long-term survival of a majority of melanomas that harbor these two mutations.
Keywords: BRAF mutations; ERK-MAPK pathway; TERT promoter mutations; cancer; telomerase reactivation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
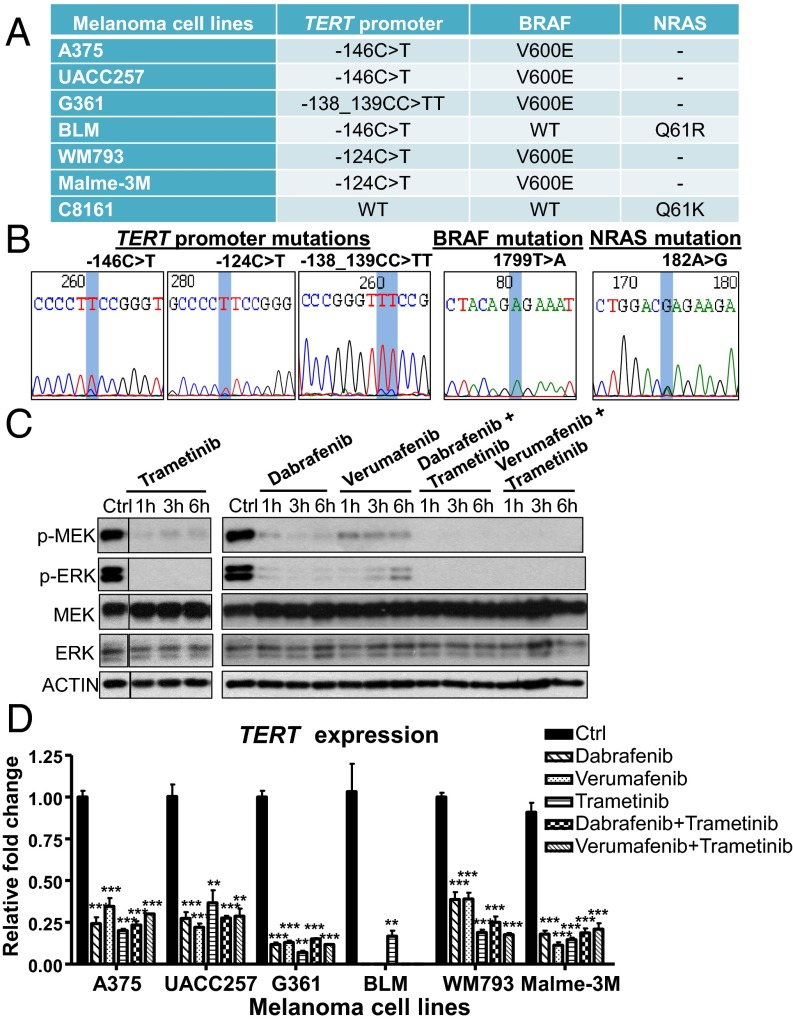
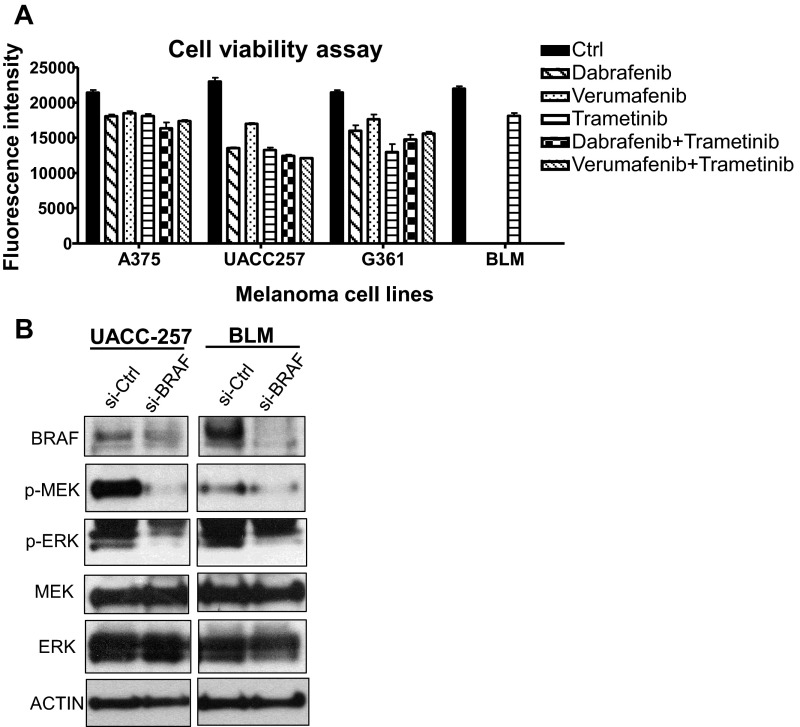
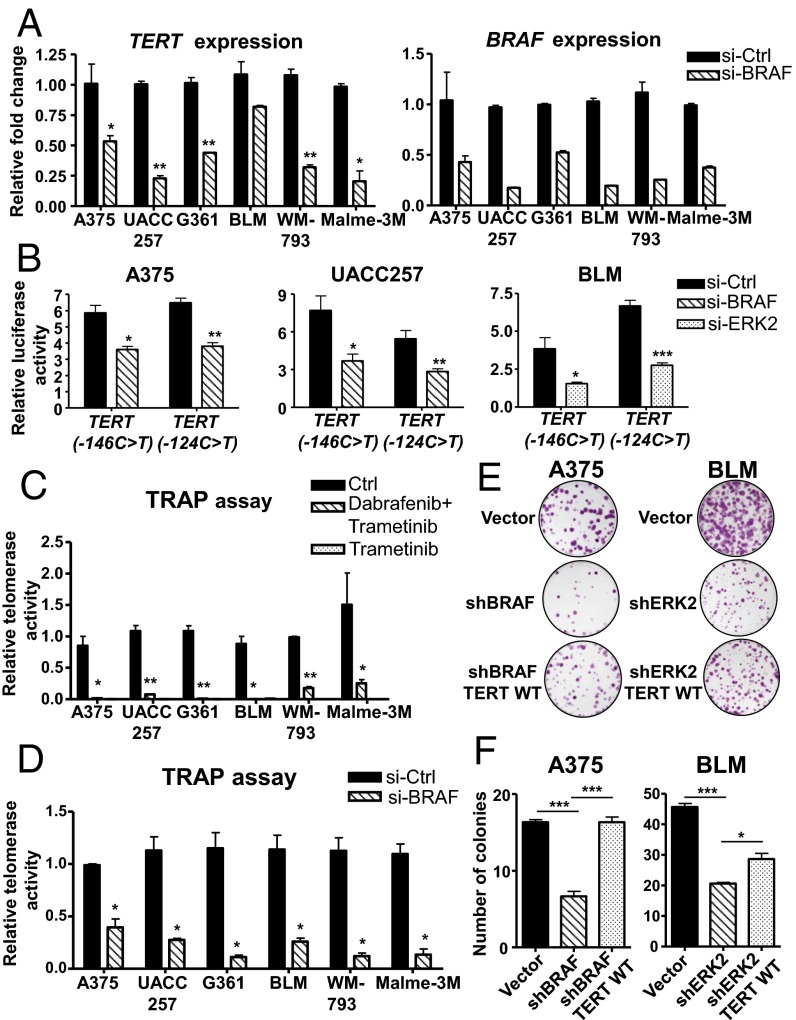
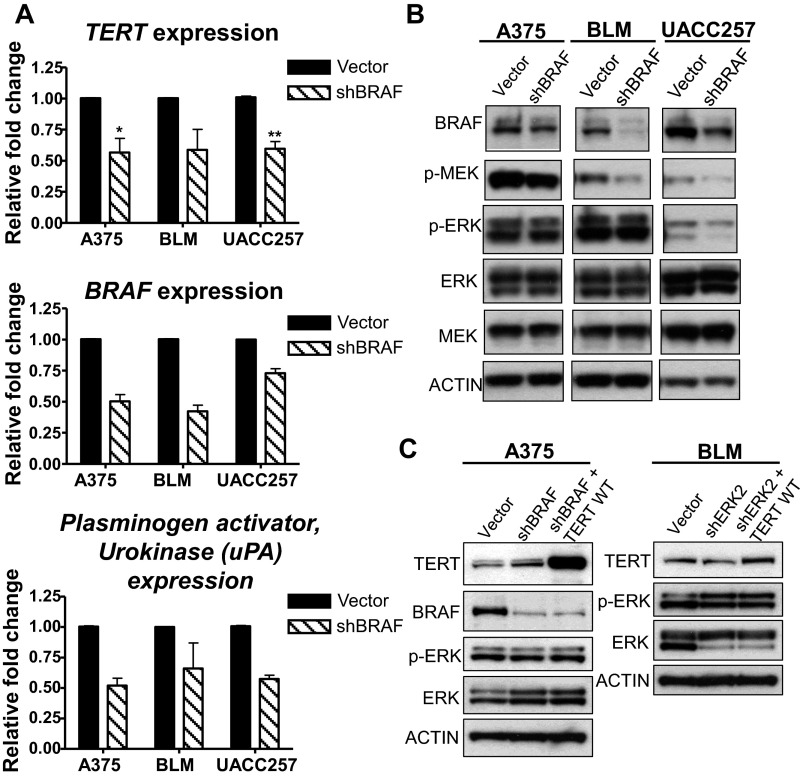
Similar articles
-
TERT promoter mutation determines apoptotic and therapeutic responses of BRAF-mutant cancers to BRAF and MEK inhibitors: Achilles Heel.Proc Natl Acad Sci U S A. 2020 Jul 7;117(27):15846-15851. doi: 10.1073/pnas.2004707117. Epub 2020 Jun 19. Proc Natl Acad Sci U S A. 2020. PMID: 32561648 Free PMC article.
-
MAPK Pathway and TERT Promoter Gene Mutation Pattern and Its Prognostic Value in Melanoma Patients: A Retrospective Study of 2,793 Cases.Clin Cancer Res. 2017 Oct 15;23(20):6120-6127. doi: 10.1158/1078-0432.CCR-17-0980. Epub 2017 Jul 18. Clin Cancer Res. 2017. PMID: 28720667
-
TERT promoter mutations in melanoma render TERT expression dependent on MAPK pathway activation.Oncotarget. 2016 Aug 16;7(33):53127-53136. doi: 10.18632/oncotarget.10634. Oncotarget. 2016. PMID: 27449293 Free PMC article.
-
Targeting BRAF in melanoma: biological and clinical challenges.Crit Rev Oncol Hematol. 2013 Sep;87(3):239-55. doi: 10.1016/j.critrevonc.2013.01.003. Epub 2013 Feb 15. Crit Rev Oncol Hematol. 2013. PMID: 23415641 Review.
-
Mitogen-Activated Protein Kinase Signaling Pathway in Cutaneous Melanoma: An Updated Review.Arch Pathol Lab Med. 2016 Nov;140(11):1290-1296. doi: 10.5858/arpa.2015-0475-RS. Arch Pathol Lab Med. 2016. PMID: 27788045 Review.
Cited by
-
Role of novel histone modifications in cancer.Oncotarget. 2017 Dec 17;9(13):11414-11426. doi: 10.18632/oncotarget.23356. eCollection 2018 Feb 16. Oncotarget. 2017. PMID: 29541423 Free PMC article. Review.
-
Melanoma: Genetic Abnormalities, Tumor Progression, Clonal Evolution and Tumor Initiating Cells.Med Sci (Basel). 2017 Nov 20;5(4):28. doi: 10.3390/medsci5040028. Med Sci (Basel). 2017. PMID: 29156643 Free PMC article. Review.
-
Non-canonical roles of canonical telomere binding proteins in cancers.Cell Mol Life Sci. 2021 May;78(9):4235-4257. doi: 10.1007/s00018-021-03783-0. Epub 2021 Feb 18. Cell Mol Life Sci. 2021. PMID: 33599797 Free PMC article. Review.
-
Fangchinoline, a Bisbenzylisoquinoline Alkaloid can Modulate Cytokine-Impelled Apoptosis via the Dual Regulation of NF-κB and AP-1 Pathways.Molecules. 2019 Aug 28;24(17):3127. doi: 10.3390/molecules24173127. Molecules. 2019. PMID: 31466313 Free PMC article.
-
Pathogenic roles of long noncoding RNAs in melanoma: Implications in diagnosis and therapies.Genes Dis. 2021 Sep 17;10(1):113-125. doi: 10.1016/j.gendis.2021.08.007. eCollection 2023 Jan. Genes Dis. 2021. PMID: 37013035 Free PMC article. Review.
References
-
- Paluncic J, et al. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim Biophys Acta. 2016;1863(4):770–784. - PubMed
-
- Poulikakos PI, Rosen N. Mutant BRAF melanomas: Dependence and resistance. Cancer Cell. 2011;19(1):11–15. - PubMed
-
- Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
