

HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62-associated proteostasis
- PMID: 27846364
- PMCID: PMC5240838
- DOI: 10.1080/15548627.2016.1248018
HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62-associated proteostasis
Abstract
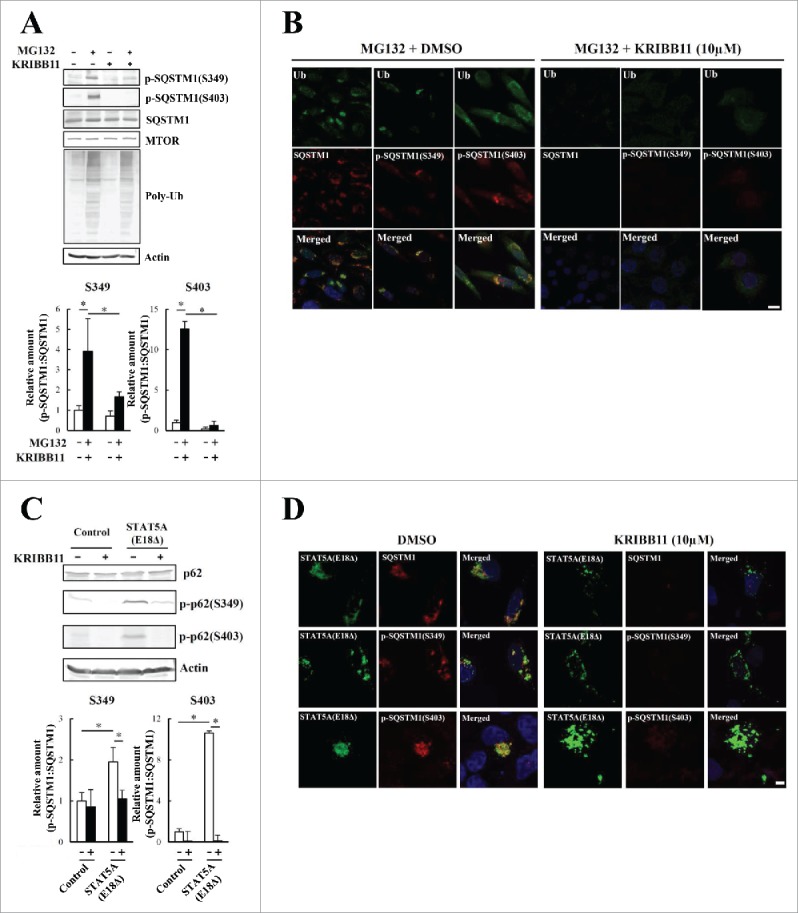
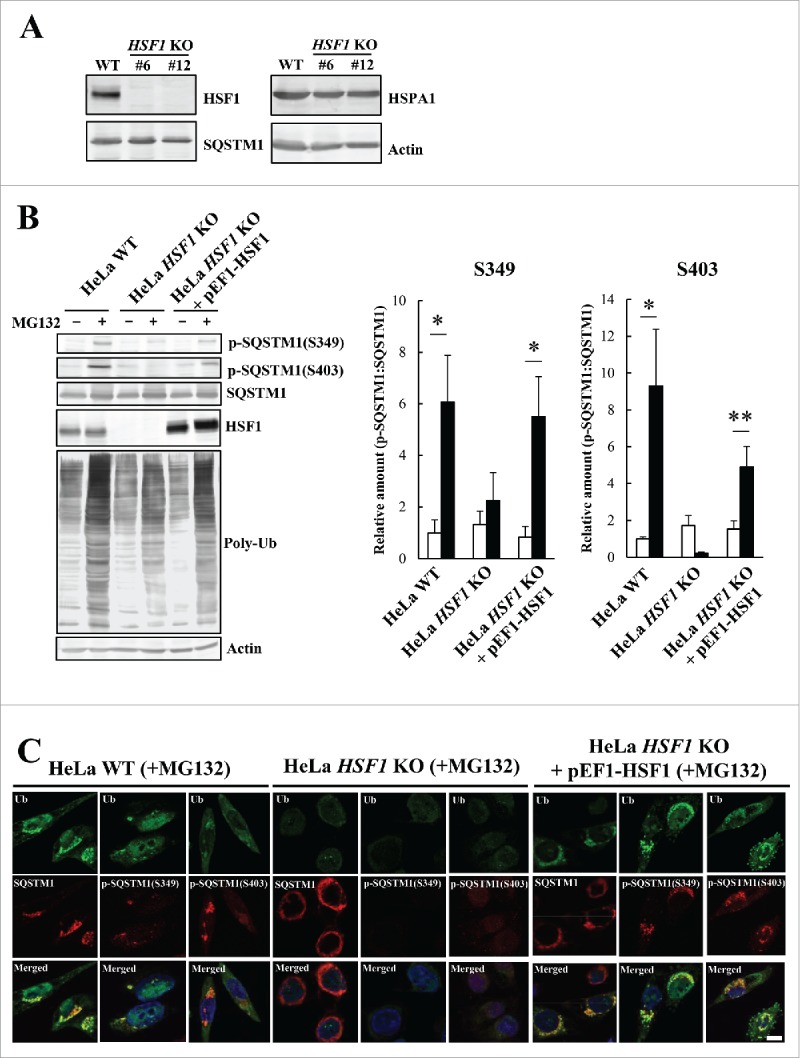
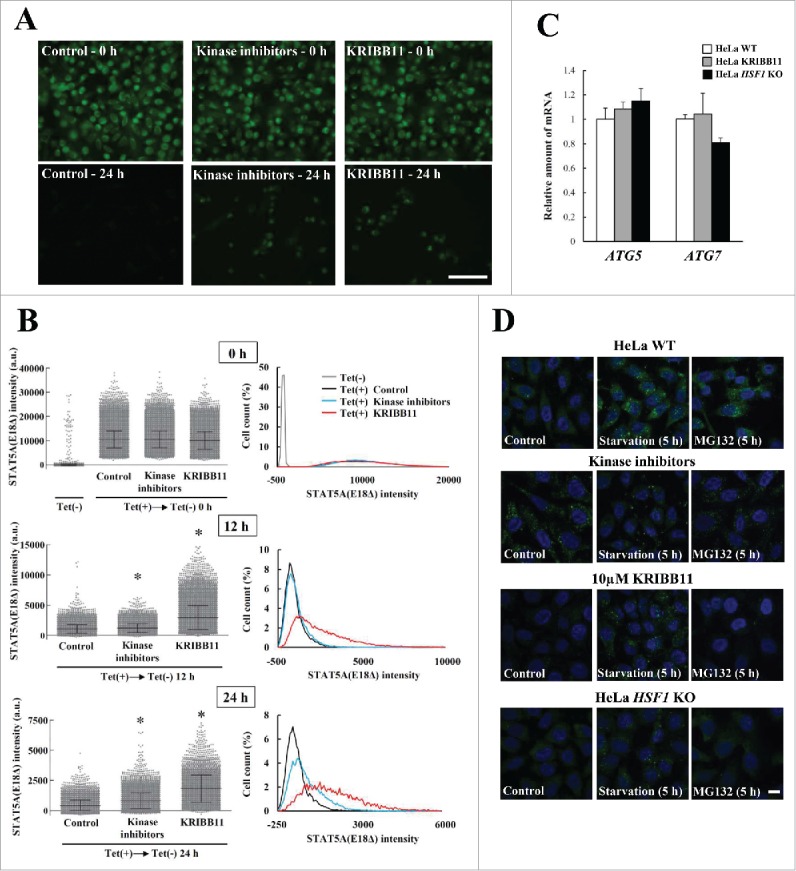
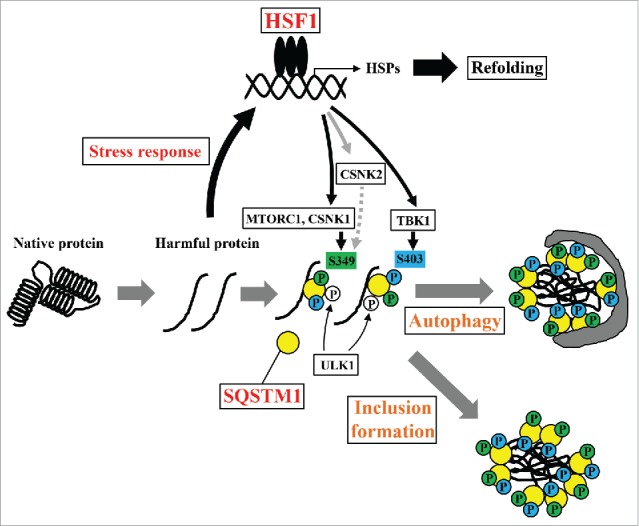
Proteostasis is important for protecting cells from harmful proteins and is mainly controlled by the HSF1 (heat shock transcription factor 1) stress response pathway. This pathway facilitates protein refolding by molecular chaperones; however, it is unclear whether it functions in autophagy or inclusion formation. The autophagy receptor SQSTM1/p62 is involved in selective autophagic clearance and inclusion formation by harmful proteins, and its phosphorylation at S349, S403, and S407 is required for binding to substrates. Here, we demonstrate that casein kinase 1 phosphorylates the SQSTM1 S349 residue when harmful proteins accumulate. Investigation of upstream factors showed that both SQSTM1 S349 and SQSTM1 S403 residues were phosphorylated in an HSF1 dependent manner. Inhibition of SQSTM1 phosphorylation suppressed inclusion formation by ubiquitinated proteins and prevented colocalization of SQSTM1 with aggregation-prone proteins. Moreover, HSF1 inhibition impaired aggregate-induced autophagosome formation and elimination of protein aggregates. Our findings indicate that HSF1 triggers SQSTM1-mediated proteostasis.
Keywords: HSF1; SQSTM1/p62; aggrephagy; casein kinase; phosphorylation; proteostasis.
Figures
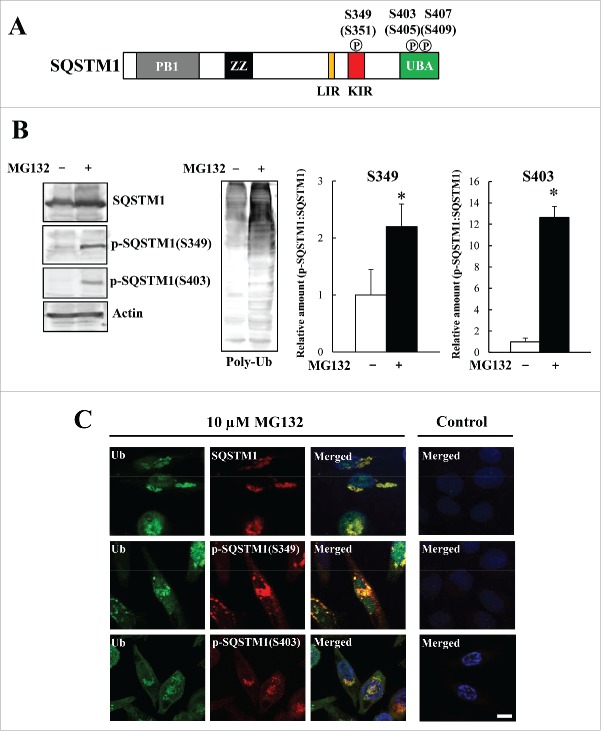
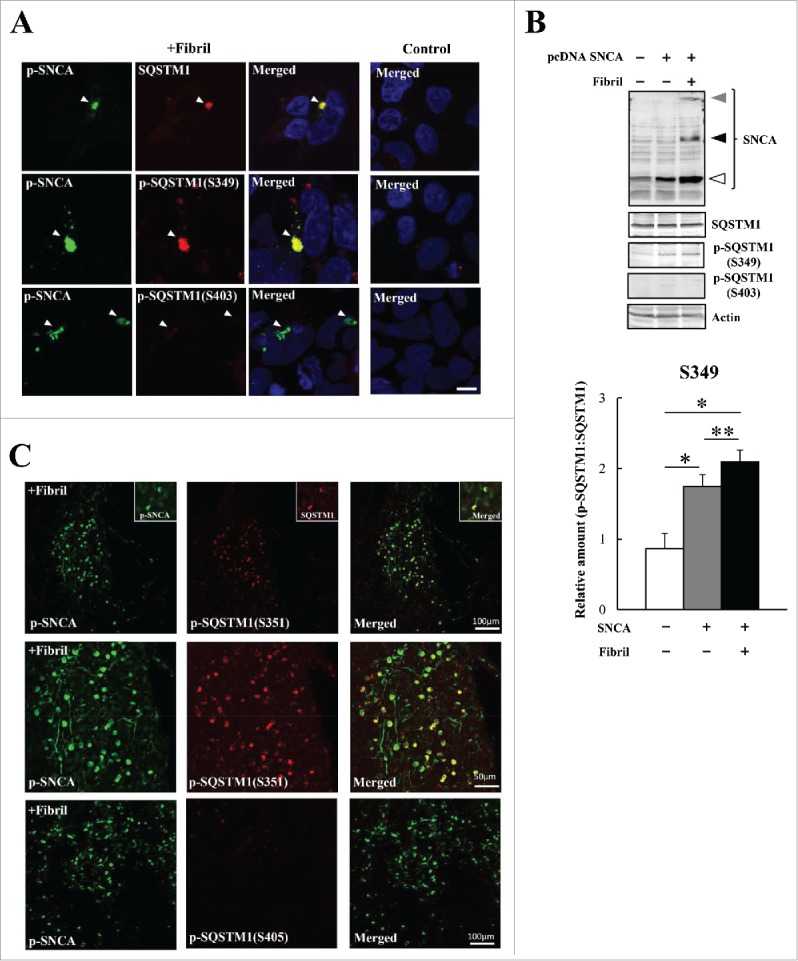
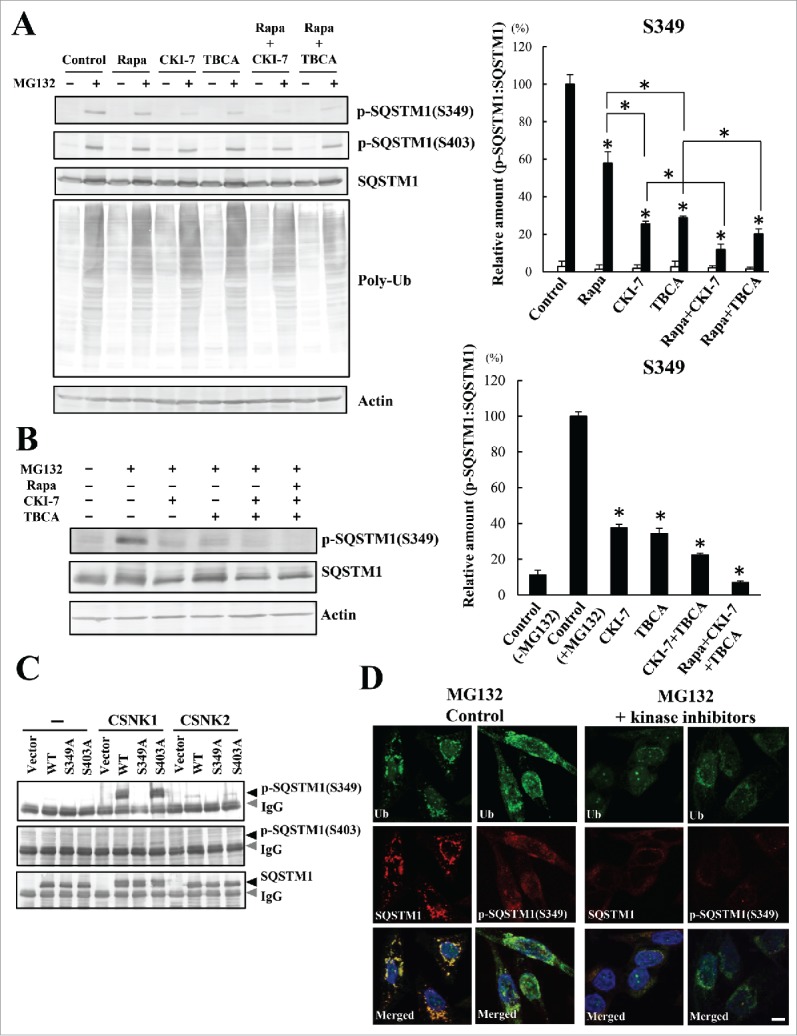
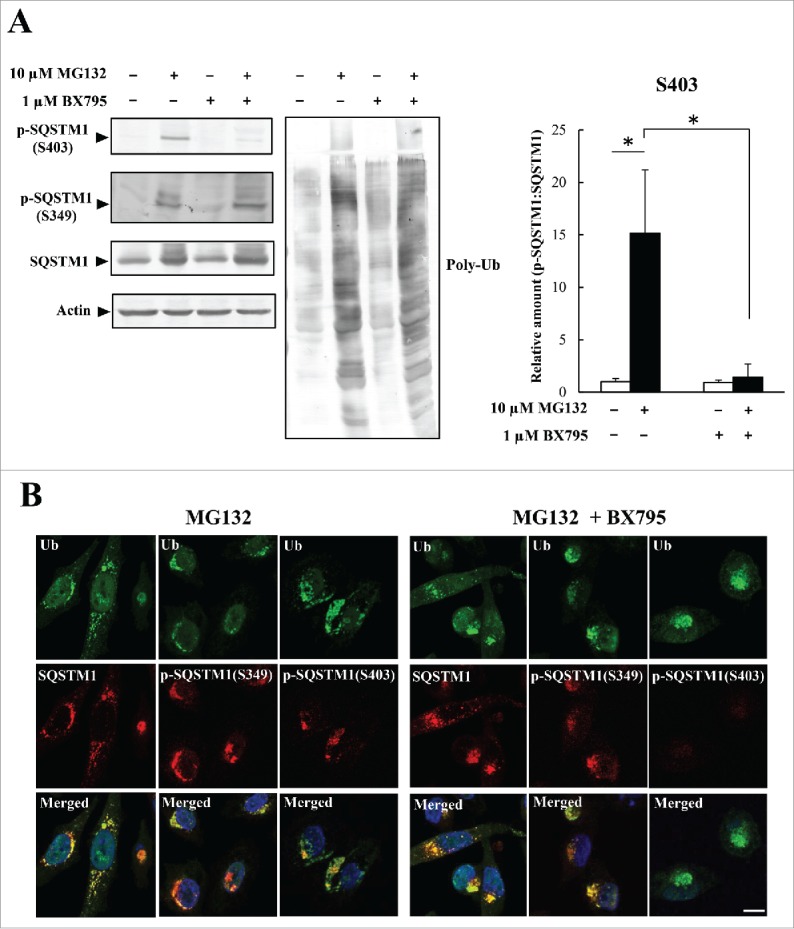
Similar articles
-
Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins.Mol Cell. 2011 Oct 21;44(2):279-89. doi: 10.1016/j.molcel.2011.07.039. Mol Cell. 2011. PMID: 22017874
-
A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells.Autophagy. 2015;11(7):1161-78. doi: 10.1080/15548627.2015.1052928. Autophagy. 2015. PMID: 26043024 Free PMC article.
-
PLK2-mediated phosphorylation of SQSTM1 S349 promotes aggregation of polyubiquitinated proteins upon proteasomal dysfunction.Autophagy. 2024 Oct;20(10):2221-2237. doi: 10.1080/15548627.2024.2361574. Epub 2024 Jun 19. Autophagy. 2024. PMID: 39316746
-
Regulation of selective autophagy: the p62/SQSTM1 paradigm.Essays Biochem. 2017 Dec 12;61(6):609-624. doi: 10.1042/EBC20170035. Print 2017 Dec 12. Essays Biochem. 2017. PMID: 29233872 Review.
-
p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation.Cell Mol Biol Lett. 2016 Dec 13;21:29. doi: 10.1186/s11658-016-0031-z. eCollection 2016. Cell Mol Biol Lett. 2016. PMID: 28536631 Free PMC article. Review.
Cited by
-
Ubiquitin, Autophagy and Neurodegenerative Diseases.Cells. 2020 Sep 2;9(9):2022. doi: 10.3390/cells9092022. Cells. 2020. PMID: 32887381 Free PMC article. Review.
-
The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy.Cells. 2019 Jan 10;8(1):40. doi: 10.3390/cells8010040. Cells. 2019. PMID: 30634694 Free PMC article. Review.
-
A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons.Cell Death Dis. 2019 Feb 12;10(2):133. doi: 10.1038/s41419-019-1364-0. Cell Death Dis. 2019. PMID: 30755581 Free PMC article.
-
Clinical disease progression and biomarkers in Niemann-Pick disease type C: a prospective cohort study.Orphanet J Rare Dis. 2020 Nov 23;15(1):328. doi: 10.1186/s13023-020-01616-0. Orphanet J Rare Dis. 2020. PMID: 33228797 Free PMC article.
-
Is amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease?Mol Neurodegener. 2017 Dec 28;12(1):90. doi: 10.1186/s13024-017-0232-6. Mol Neurodegener. 2017. PMID: 29282133 Free PMC article. Review.
References
-
- Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem 2015; 84:435-64; PMID:25784053; http://dx.doi.org/10.1146/annurev-biochem-060614-033955 - DOI - PMC - PubMed
-
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 1998; 12:3788-96; PMID:9869631; http://dx.doi.org/10.1101/gad.12.24.3788 - DOI - PubMed
-
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780-6; PMID:17051204; http://dx.doi.org/10.1038/nature05291 - DOI - PubMed
-
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/10.4161/auto.7.3.14487 - DOI - PMC - PubMed
-
- Knaevelsrud H, Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett 2010; 584:2635-45; PMID:20412801; http://dx.doi.org/10.1016/j.febslet.2010.04.041 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials