

MECR Mutations Cause Childhood-Onset Dystonia and Optic Atrophy, a Mitochondrial Fatty Acid Synthesis Disorder
- PMID: 27817865
- PMCID: PMC5142118
- DOI: 10.1016/j.ajhg.2016.09.021
MECR Mutations Cause Childhood-Onset Dystonia and Optic Atrophy, a Mitochondrial Fatty Acid Synthesis Disorder
Abstract
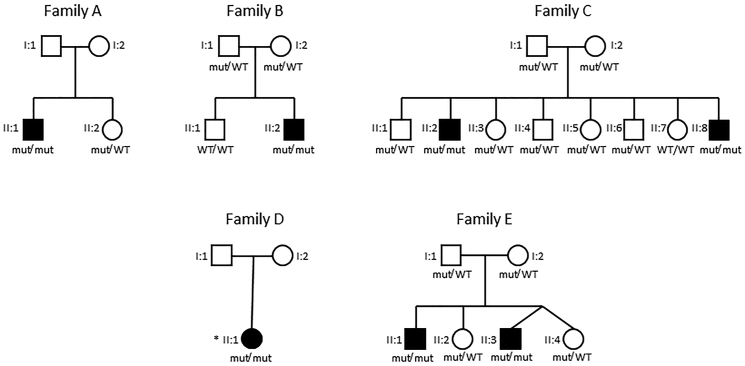
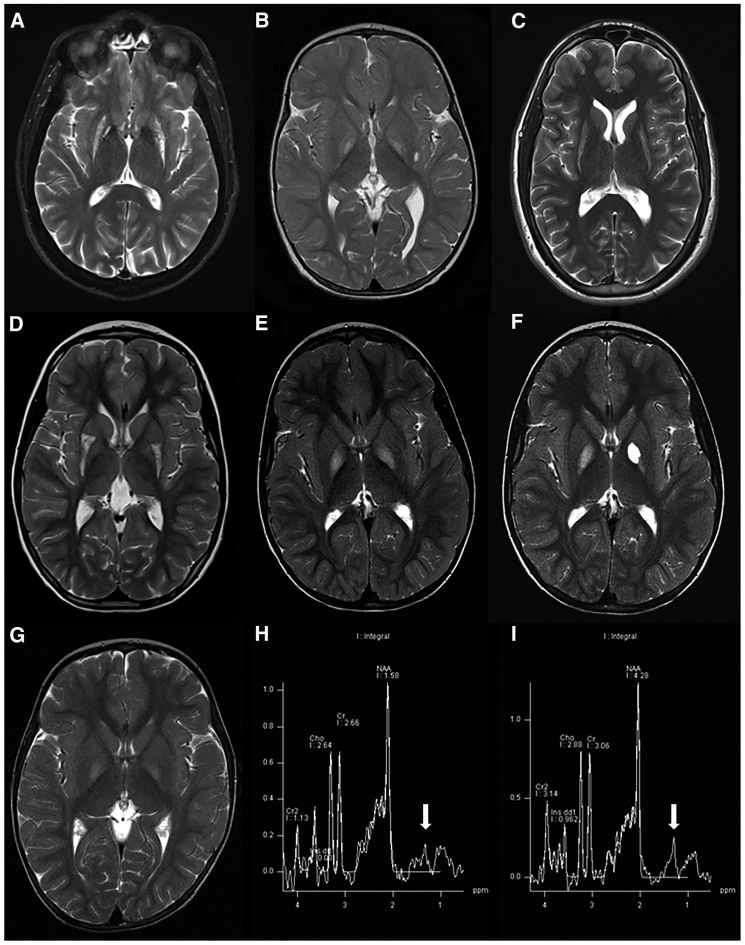
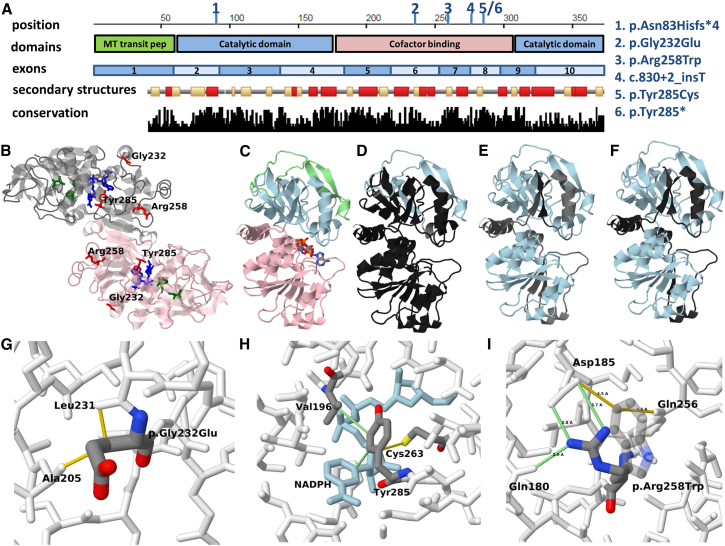
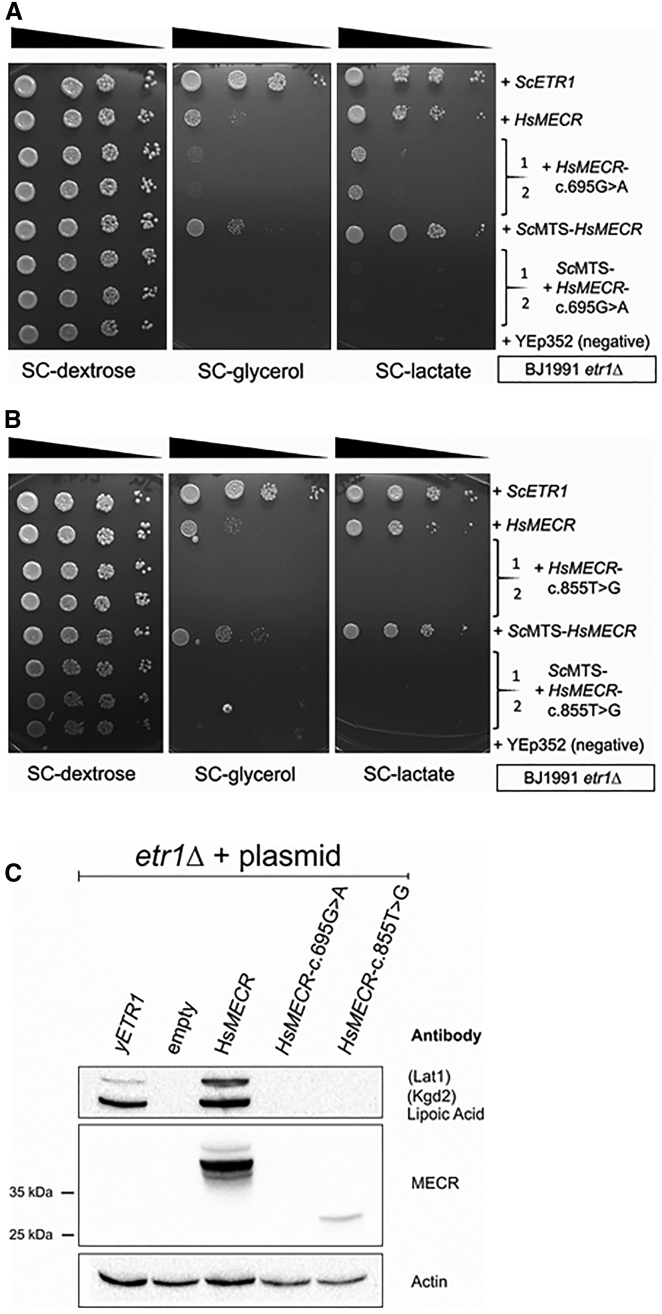
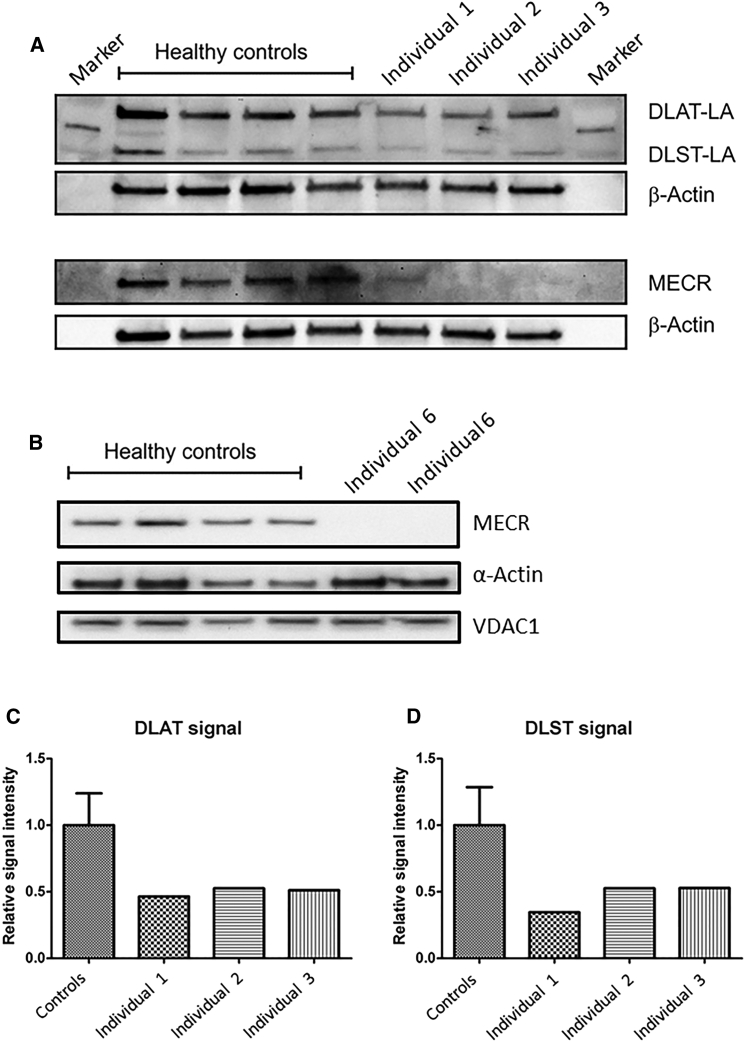
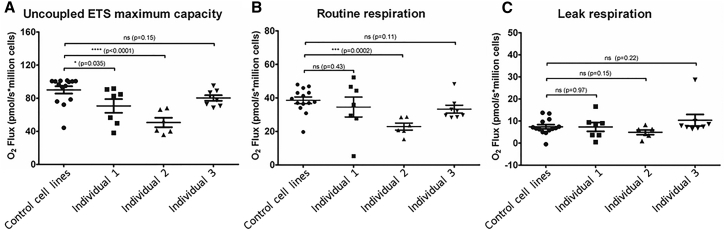
Mitochondrial fatty acid synthesis (mtFAS) is an evolutionarily conserved pathway essential for the function of the respiratory chain and several mitochondrial enzyme complexes. We report here a unique neurometabolic human disorder caused by defective mtFAS. Seven individuals from five unrelated families presented with childhood-onset dystonia, optic atrophy, and basal ganglia signal abnormalities on MRI. All affected individuals were found to harbor recessive mutations in MECR encoding the mitochondrial trans-2-enoyl-coenzyme A-reductase involved in human mtFAS. All six mutations are extremely rare in the general population, segregate with the disease in the families, and are predicted to be deleterious. The nonsense c.855T>G (p.Tyr285∗), c.247_250del (p.Asn83Hisfs∗4), and splice site c.830+2_830+3insT mutations lead to C-terminal truncation variants of MECR. The missense c.695G>A (p.Gly232Glu), c.854A>G (p.Tyr285Cys), and c.772C>T (p.Arg258Trp) mutations involve conserved amino acid residues, are located within the cofactor binding domain, and are predicted by structural analysis to have a destabilizing effect. Yeast modeling and complementation studies validated the pathogenicity of the MECR mutations. Fibroblast cell lines from affected individuals displayed reduced levels of both MECR and lipoylated proteins as well as defective respiration. These results suggest that mutations in MECR cause a distinct human disorder of the mtFAS pathway. The observation of decreased lipoylation raises the possibility of a potential therapeutic strategy.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Impaired Mitochondrial Fatty Acid Synthesis Leads to Neurodegeneration in Mice.J Neurosci. 2018 Nov 7;38(45):9781-9800. doi: 10.1523/JNEUROSCI.3514-17.2018. Epub 2018 Sep 28. J Neurosci. 2018. PMID: 30266742 Free PMC article.
-
Whole exome sequencing identifies a novel homozygous MECR mutation in a Chinese patient with childhood-onset dystonia and basal ganglia abnormalities, without optic atrophy.Mitochondrion. 2021 Mar;57:222-229. doi: 10.1016/j.mito.2020.12.014. Epub 2021 Jan 2. Mitochondrion. 2021. PMID: 33401012
-
Genetic modifications of Mecr reveal a role for mitochondrial 2-enoyl-CoA/ACP reductase in placental development in mice.Hum Mol Genet. 2017 Jun 1;26(11):2104-2117. doi: 10.1093/hmg/ddx105. Hum Mol Genet. 2017. PMID: 28369354
-
Mutations in the mitochondrial complex I assembly factor NDUFAF6 cause isolated bilateral striatal necrosis and progressive dystonia in childhood.Mol Genet Metab. 2019 Mar;126(3):250-258. doi: 10.1016/j.ymgme.2019.01.001. Epub 2019 Jan 5. Mol Genet Metab. 2019. PMID: 30642748 Review.
-
Mitochondrial Fatty Acids and Neurodegenerative Disorders.Neuroscientist. 2021 Apr;27(2):143-158. doi: 10.1177/1073858420936162. Epub 2020 Jul 9. Neuroscientist. 2021. PMID: 32644907 Review.
Cited by
-
Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria.Elife. 2020 Aug 17;9:e58041. doi: 10.7554/eLife.58041. Elife. 2020. PMID: 32804083 Free PMC article.
-
Impact of Mitochondrial Fatty Acid Synthesis on Mitochondrial Biogenesis.Curr Biol. 2018 Oct 22;28(20):R1212-R1219. doi: 10.1016/j.cub.2018.08.022. Curr Biol. 2018. PMID: 30352195 Free PMC article. Review.
-
Recessive pathogenic variants in MCAT cause combined oxidative phosphorylation deficiency.Elife. 2023 Mar 7;12:e68047. doi: 10.7554/eLife.68047. Elife. 2023. PMID: 36881526 Free PMC article.
-
Brain metabolism and neurological symptoms in combined malonic and methylmalonic aciduria.Orphanet J Rare Dis. 2020 Jan 22;15(1):27. doi: 10.1186/s13023-020-1299-7. Orphanet J Rare Dis. 2020. PMID: 31969167 Free PMC article.
-
Protein moonlighting elucidates the essential human pathway catalyzing lipoic acid assembly on its cognate enzymes.Proc Natl Acad Sci U S A. 2018 Jul 24;115(30):E7063-E7072. doi: 10.1073/pnas.1805862115. Epub 2018 Jul 9. Proc Natl Acad Sci U S A. 2018. PMID: 29987032 Free PMC article.
References
-
- Schneider S.A., Bhatia K.P. Secondary dystonia--clinical clues and syndromic associations. Eur. J. Neurol. 2010;17(Suppl 1):52–57. - PubMed
-
- Zuccoli G., Yannes M.P., Nardone R., Bailey A., Goldstein A. Bilateral symmetrical basal ganglia and thalamic lesions in children: an update (2015) Neuroradiology. 2015;57:973–989. - PubMed
-
- Meyer E., Kurian M.A., Hayflick S.J. Neurodegeneration with brain iron accumulation: genetic diversity and pathophysiological mechanisms. Annu. Rev. Genomics Hum. Genet. 2015;16:257–279. - PubMed
-
- Lake N.J., Compton A.G., Rahman S., Thorburn D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016;79:190–203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
