

A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns
- PMID: 27732578
- PMCID: PMC5446075
- DOI: 10.1038/nature19823
A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns
Erratum in
-
Erratum: A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns.Nature. 2017 Feb 2;542(7639):124. doi: 10.1038/nature20164. Epub 2016 Nov 16. Nature. 2017. PMID: 27851734 No abstract available.
Abstract
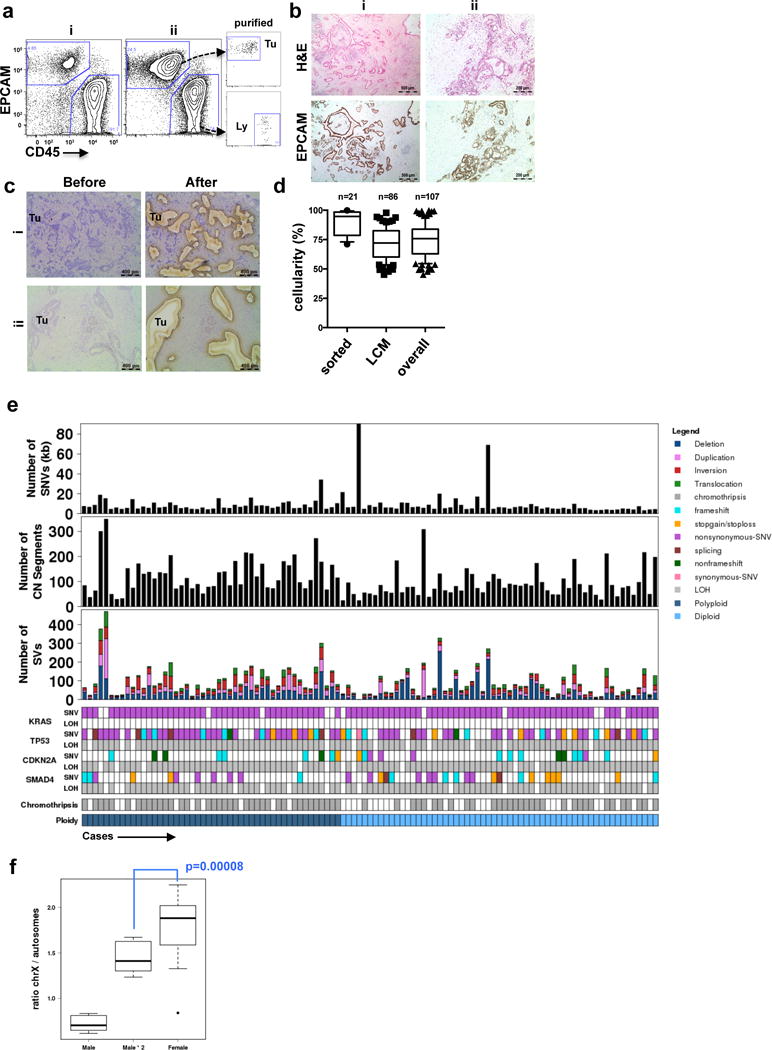
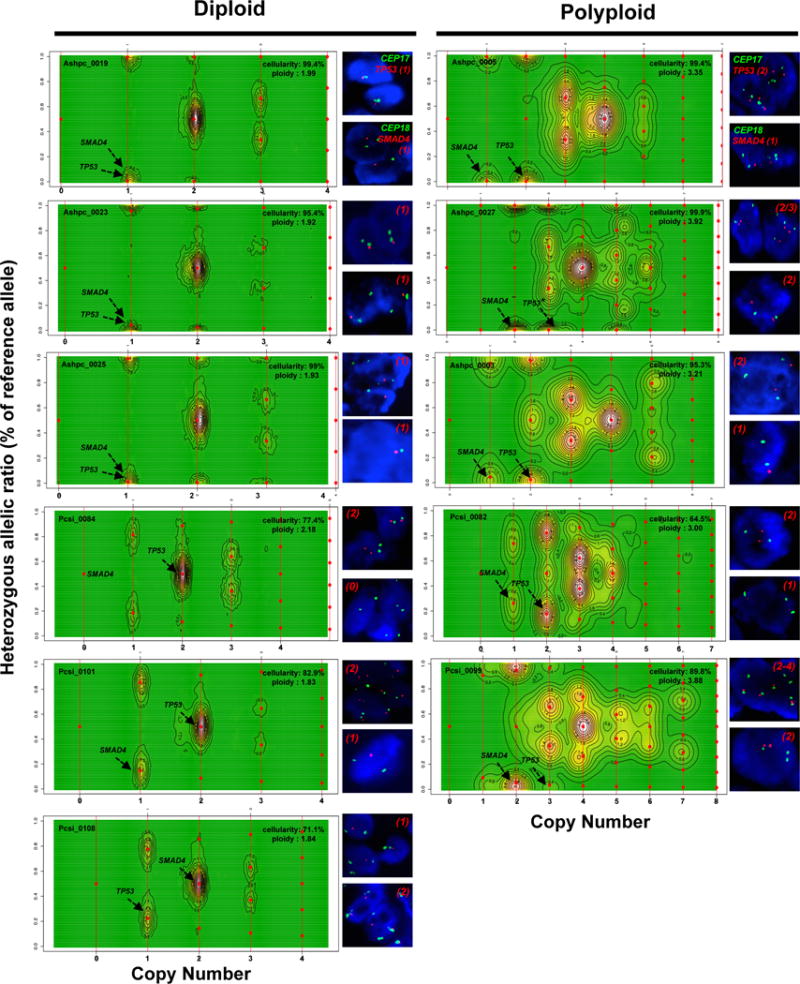
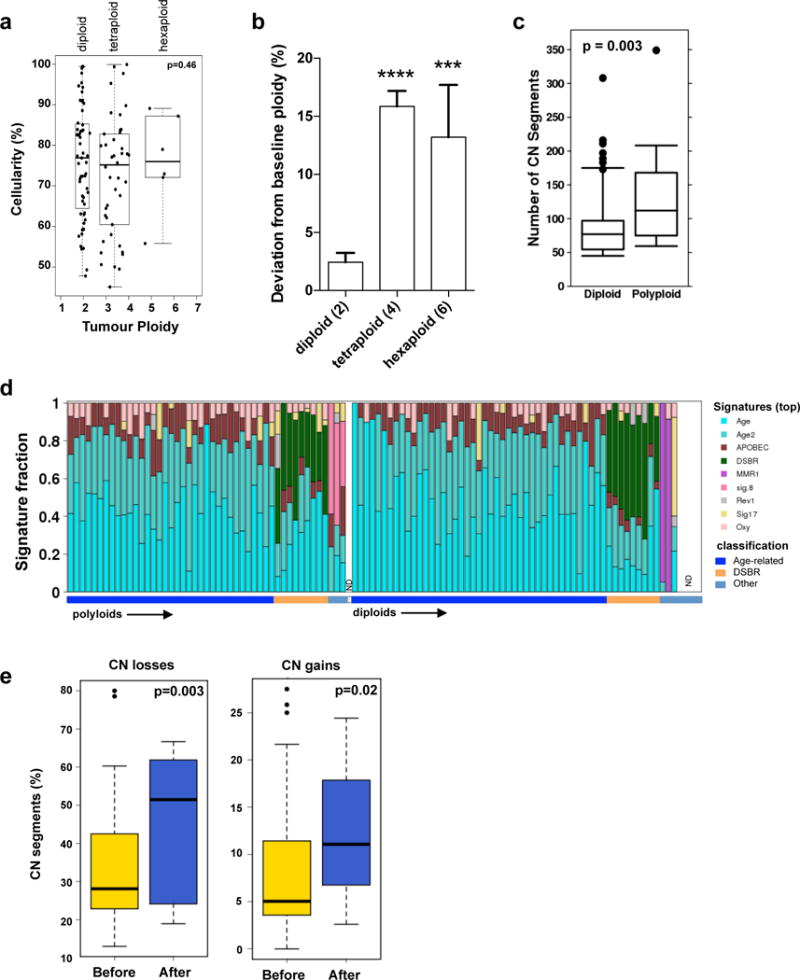
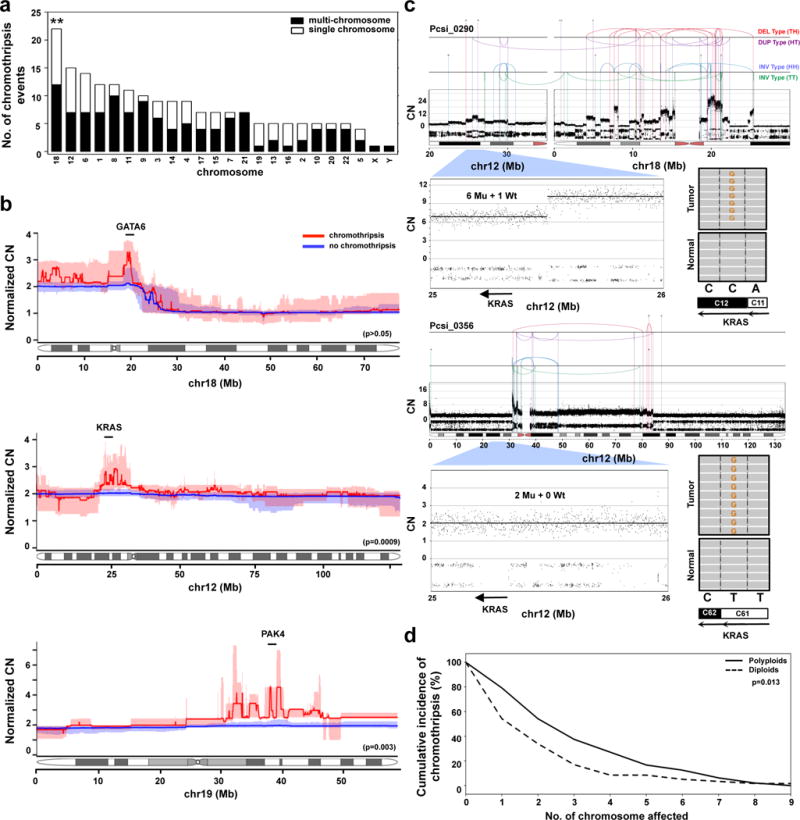
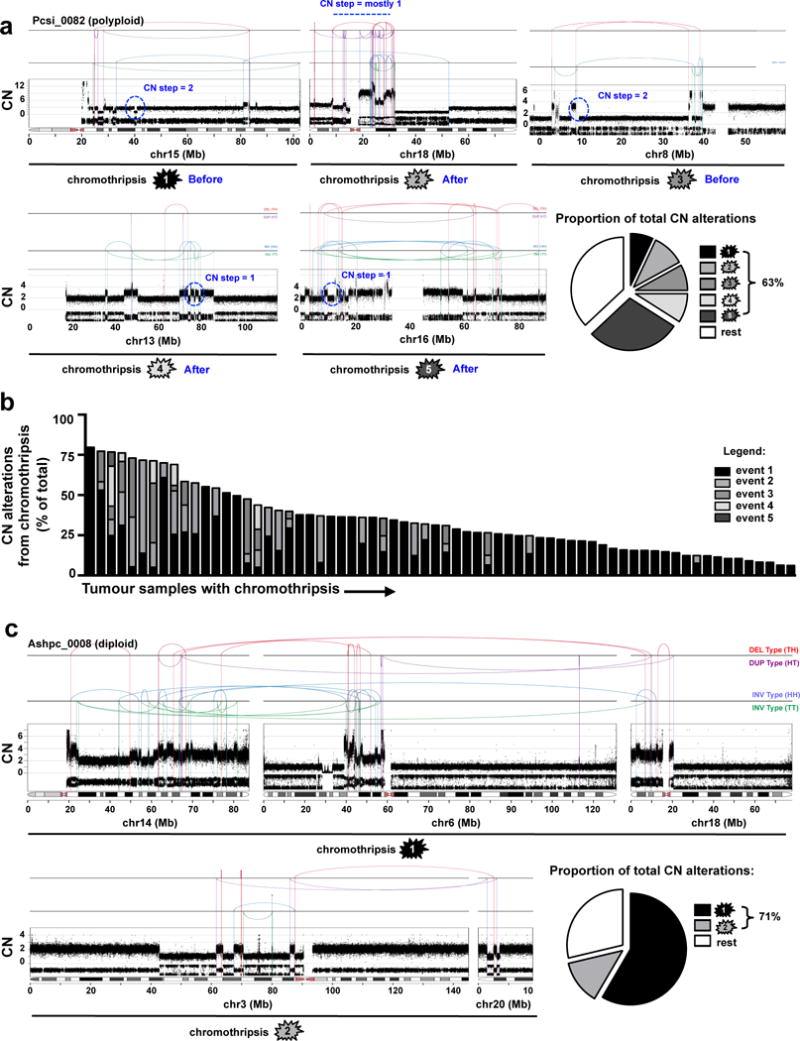
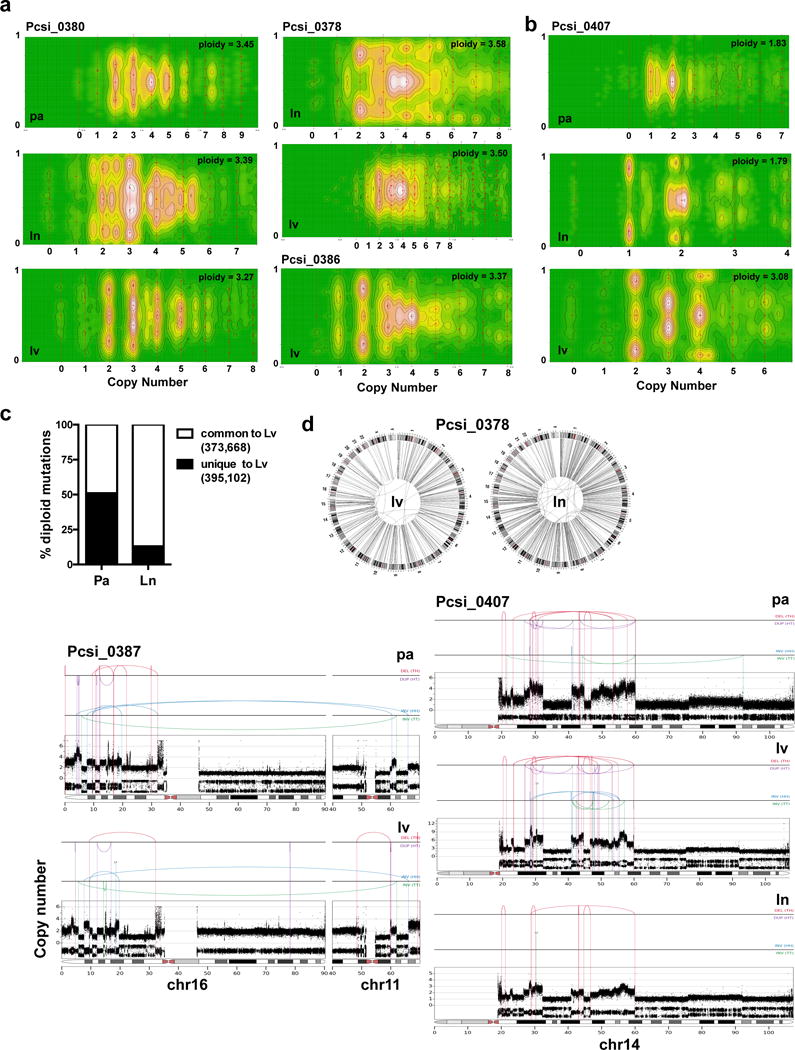
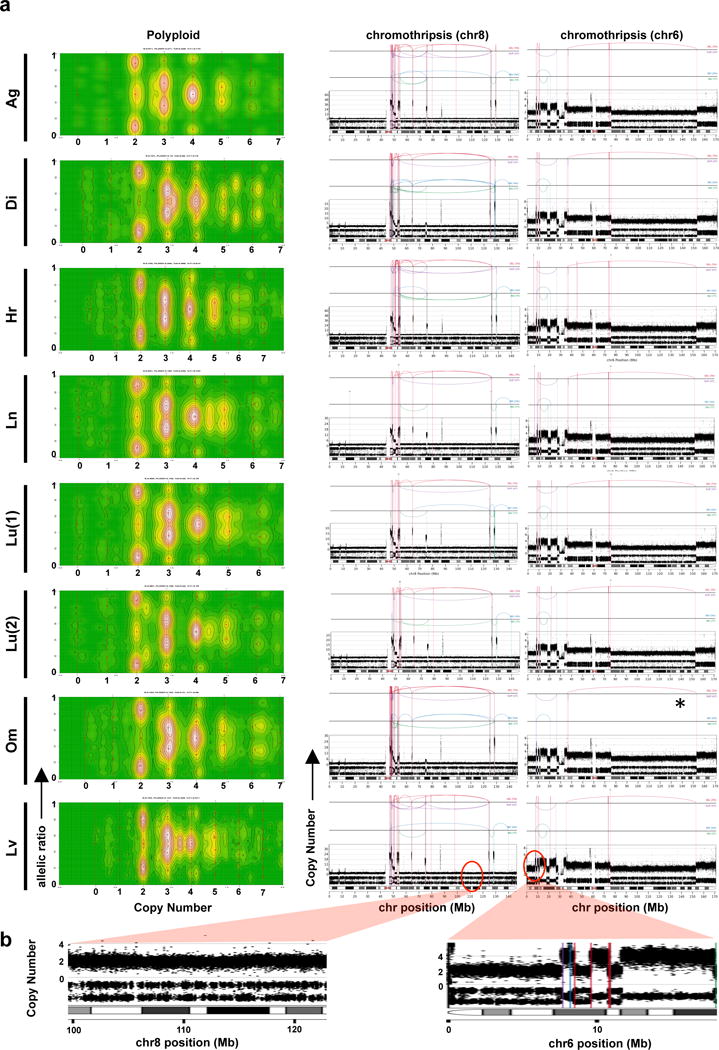
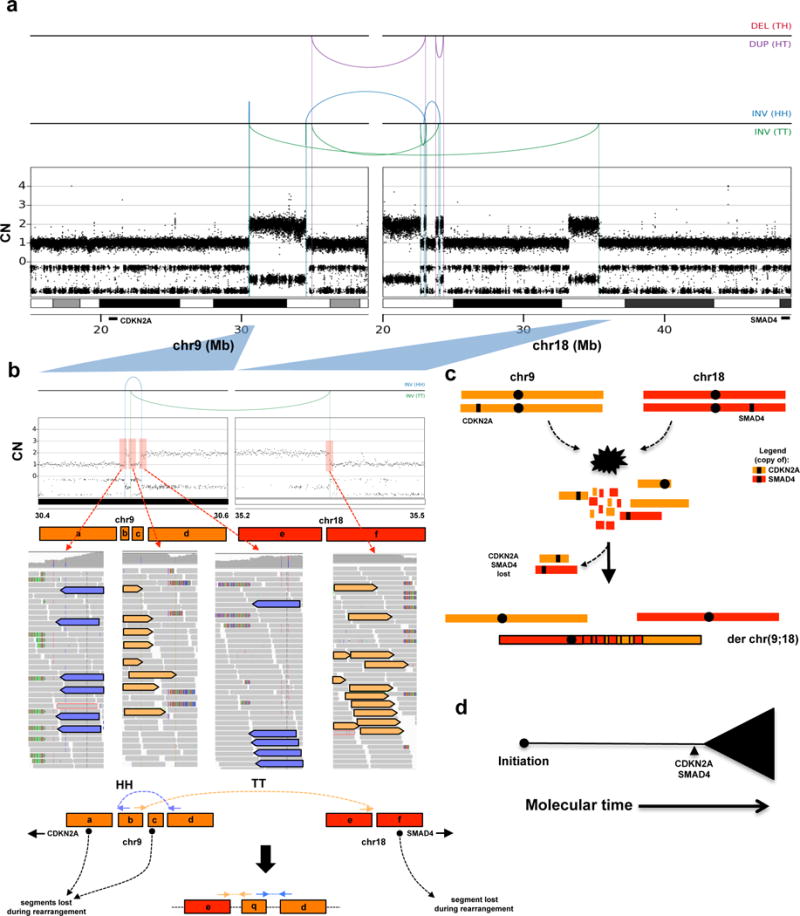
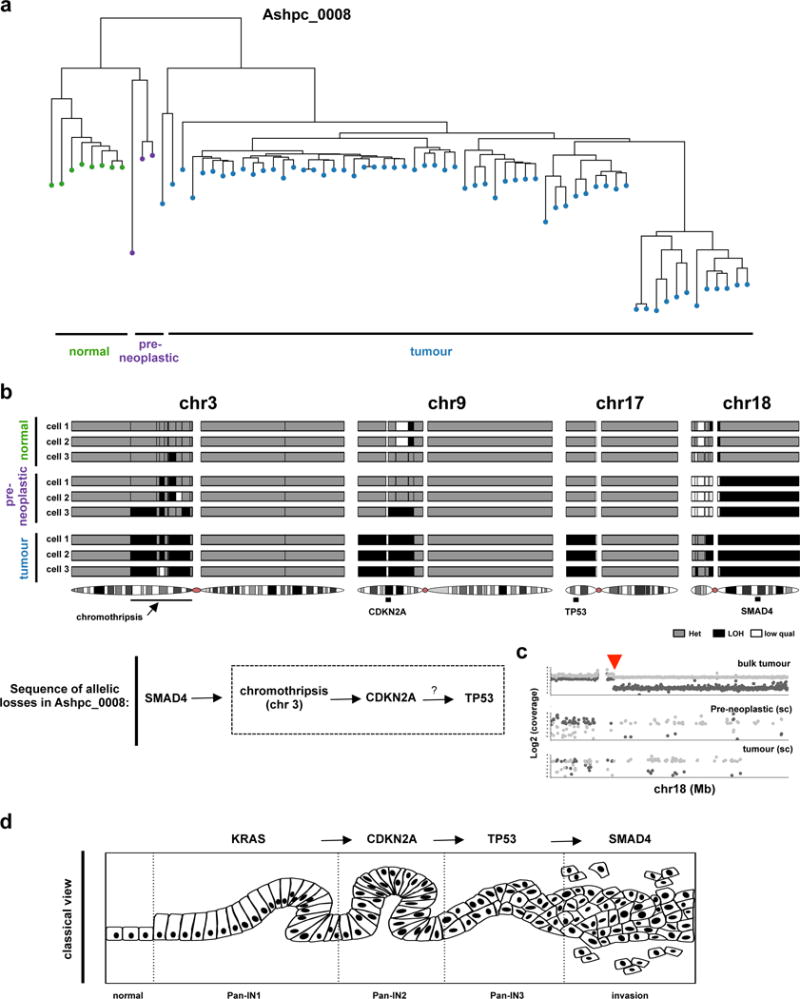
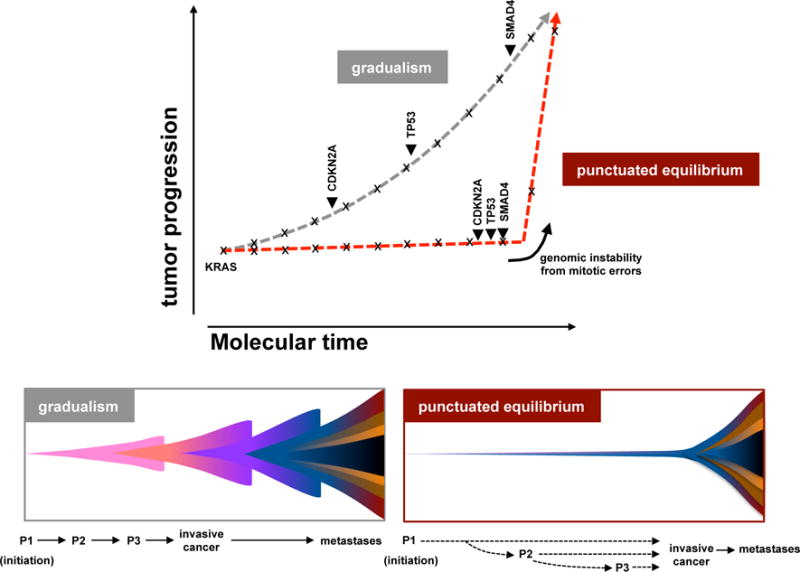
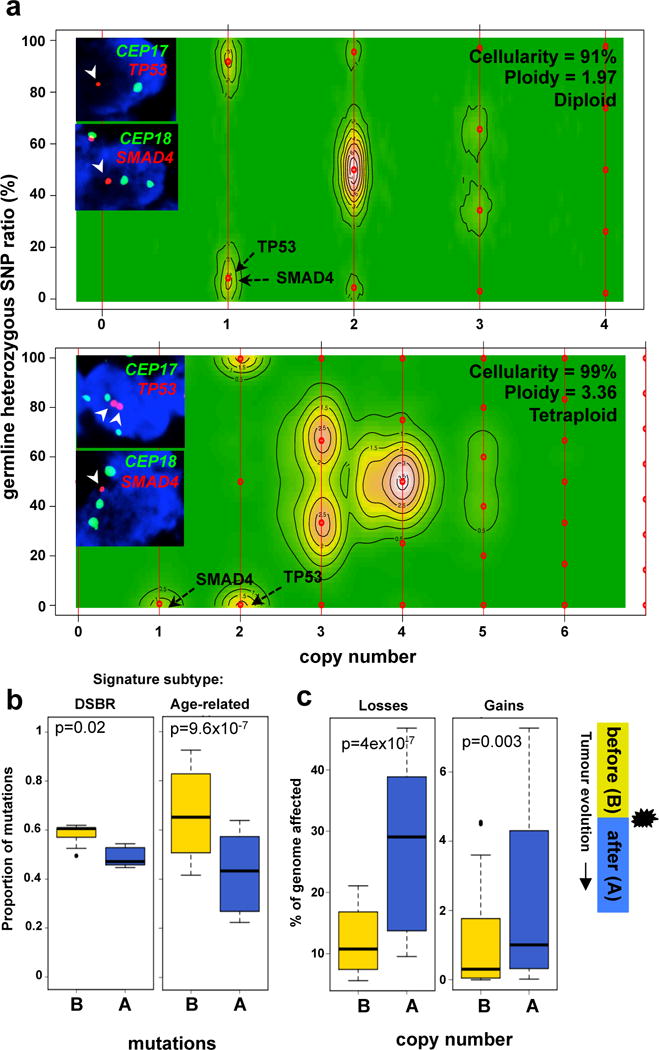
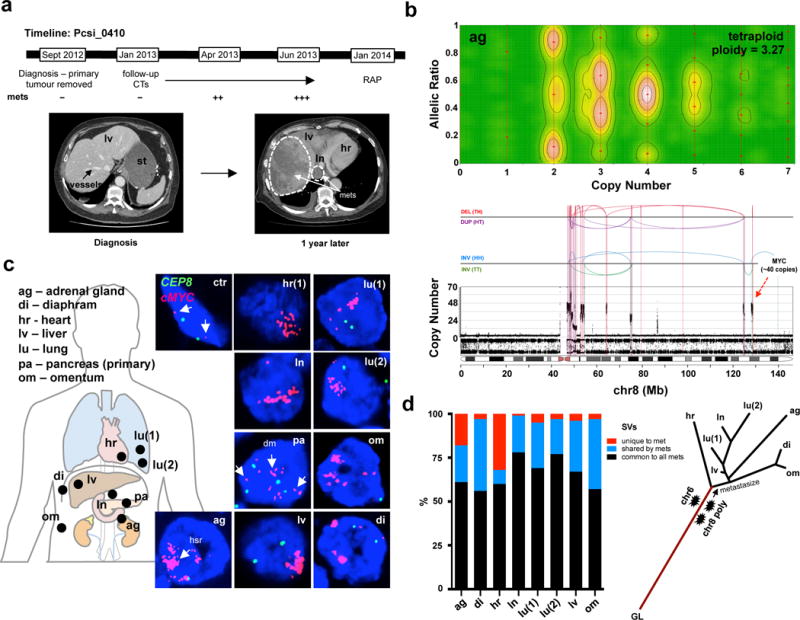
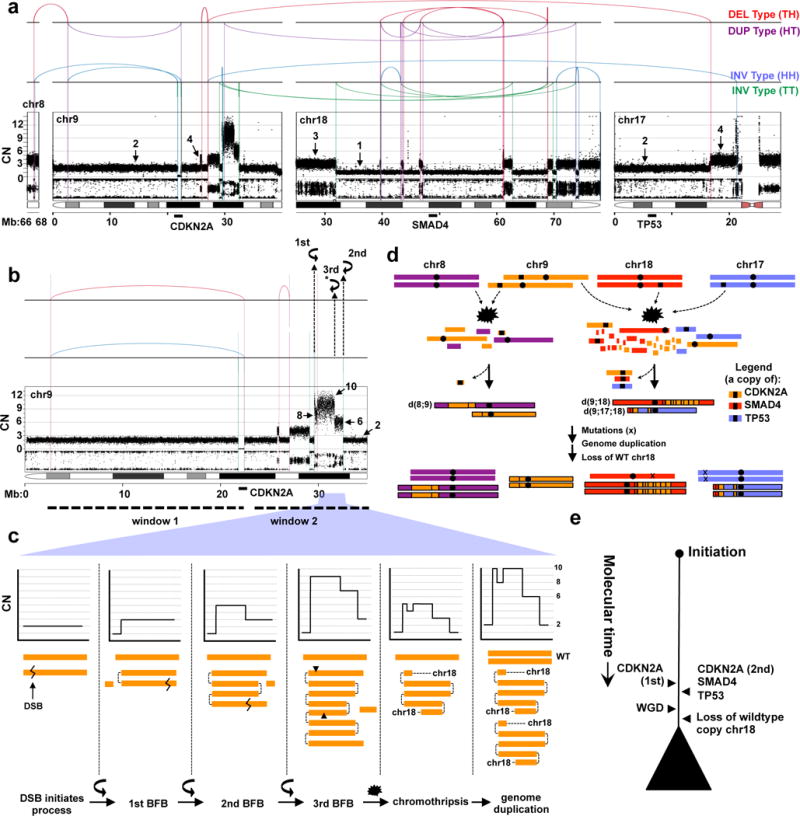
Pancreatic cancer, a highly aggressive tumour type with uniformly poor prognosis, exemplifies the classically held view of stepwise cancer development. The current model of tumorigenesis, based on analyses of precursor lesions, termed pancreatic intraepithelial neoplasm (PanINs) lesions, makes two predictions: first, that pancreatic cancer develops through a particular sequence of genetic alterations (KRAS, followed by CDKN2A, then TP53 and SMAD4); and second, that the evolutionary trajectory of pancreatic cancer progression is gradual because each alteration is acquired independently. A shortcoming of this model is that clonally expanded precursor lesions do not always belong to the tumour lineage, indicating that the evolutionary trajectory of the tumour lineage and precursor lesions can be divergent. This prevailing model of tumorigenesis has contributed to the clinical notion that pancreatic cancer evolves slowly and presents at a late stage. However, the propensity for this disease to rapidly metastasize and the inability to improve patient outcomes, despite efforts aimed at early detection, suggest that pancreatic cancer progression is not gradual. Here, using newly developed informatics tools, we tracked changes in DNA copy number and their associated rearrangements in tumour-enriched genomes and found that pancreatic cancer tumorigenesis is neither gradual nor follows the accepted mutation order. Two-thirds of tumours harbour complex rearrangement patterns associated with mitotic errors, consistent with punctuated equilibrium as the principal evolutionary trajectory. In a subset of cases, the consequence of such errors is the simultaneous, rather than sequential, knockout of canonical preneoplastic genetic drivers that are likely to set-off invasive cancer growth. These findings challenge the current progression model of pancreatic cancer and provide insights into the mutational processes that give rise to these aggressive tumours.
Figures
Comment in
-
Pancreatic cancer: Pancreatic carcinogenesis - several small steps or one giant leap?Nat Rev Gastroenterol Hepatol. 2016 Dec 22;14(1):7-8. doi: 10.1038/nrgastro.2016.190. Nat Rev Gastroenterol Hepatol. 2016. PMID: 28003666 No abstract available.
Similar articles
-
Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor.Gastroenterology. 2013 Nov;145(5):1098-1109.e1. doi: 10.1053/j.gastro.2013.07.049. Epub 2013 Aug 2. Gastroenterology. 2013. PMID: 23912084 Free PMC article.
-
Genome-Wide Somatic Copy Number Alterations and Mutations in High-Grade Pancreatic Intraepithelial Neoplasia.Am J Pathol. 2018 Jul;188(7):1723-1733. doi: 10.1016/j.ajpath.2018.03.012. Epub 2018 Apr 22. Am J Pathol. 2018. PMID: 29684357 Free PMC article.
-
The patterns and dynamics of genomic instability in metastatic pancreatic cancer.Nature. 2010 Oct 28;467(7319):1109-13. doi: 10.1038/nature09460. Nature. 2010. PMID: 20981101 Free PMC article.
-
A genetic roadmap of pancreatic cancer: still evolving.Gut. 2017 Dec;66(12):2170-2178. doi: 10.1136/gutjnl-2016-313317. Epub 2017 Oct 9. Gut. 2017. PMID: 28993418 Review.
-
From somatic mutation to early detection: insights from molecular characterization of pancreatic cancer precursor lesions.J Pathol. 2018 Dec;246(4):395-404. doi: 10.1002/path.5154. J Pathol. 2018. PMID: 30105857 Free PMC article. Review.
Cited by
-
Expandable and reversible copy number amplification drives rapid adaptation to antifungal drugs.Elife. 2020 Jul 20;9:e58349. doi: 10.7554/eLife.58349. Elife. 2020. PMID: 32687060 Free PMC article.
-
Chromothripsis is a novel biomarker for prognosis and differentiation diagnosis of pancreatic neuroendocrine neoplasms.MedComm (2020). 2024 Jul 10;5(7):e623. doi: 10.1002/mco2.623. eCollection 2024 Jul. MedComm (2020). 2024. PMID: 38988495 Free PMC article.
-
Genomic characterization of malignant progression in neoplastic pancreatic cysts.Nat Commun. 2020 Aug 14;11(1):4085. doi: 10.1038/s41467-020-17917-8. Nat Commun. 2020. PMID: 32796935 Free PMC article.
-
APOBEC3A drives deaminase domain-independent chromosomal instability to promote pancreatic cancer metastasis.Nat Cancer. 2021 Dec;2(12):1338-1356. doi: 10.1038/s43018-021-00268-8. Epub 2021 Nov 18. Nat Cancer. 2021. PMID: 35121902
-
Proteome alterations in pancreatic ductal adenocarcinoma.Cancer Lett. 2020 Jan 28;469:429-436. doi: 10.1016/j.canlet.2019.11.020. Epub 2019 Nov 14. Cancer Lett. 2020. PMID: 31734355 Free PMC article. Review.
References
-
- Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2000;6:2969–2972. - PubMed
-
- Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer research. 1997;57:2140–2143. - PubMed
-
- Wilentz RE, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer research. 1998;58:4740–4744. - PubMed
-
- Wilentz RE, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer research. 2000;60:2002–2006. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
