

Targeting CAG repeat RNAs reduces Huntington's disease phenotype independently of huntingtin levels
- PMID: 27721240
- PMCID: PMC5096913
- DOI: 10.1172/JCI83185
Targeting CAG repeat RNAs reduces Huntington's disease phenotype independently of huntingtin levels
Abstract
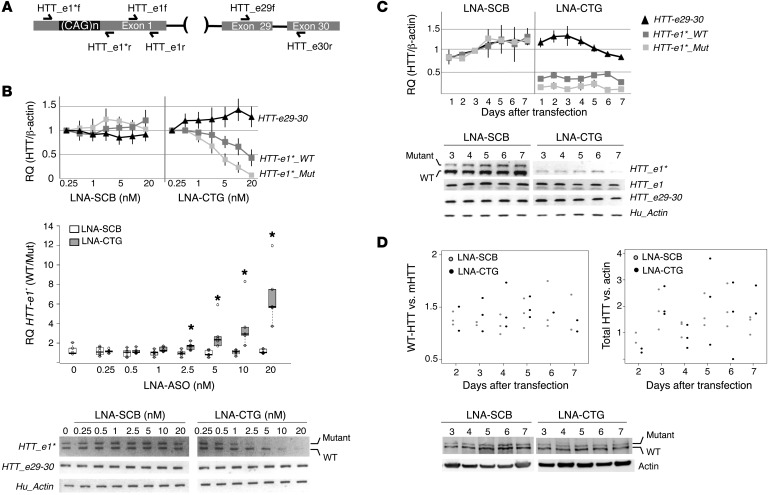
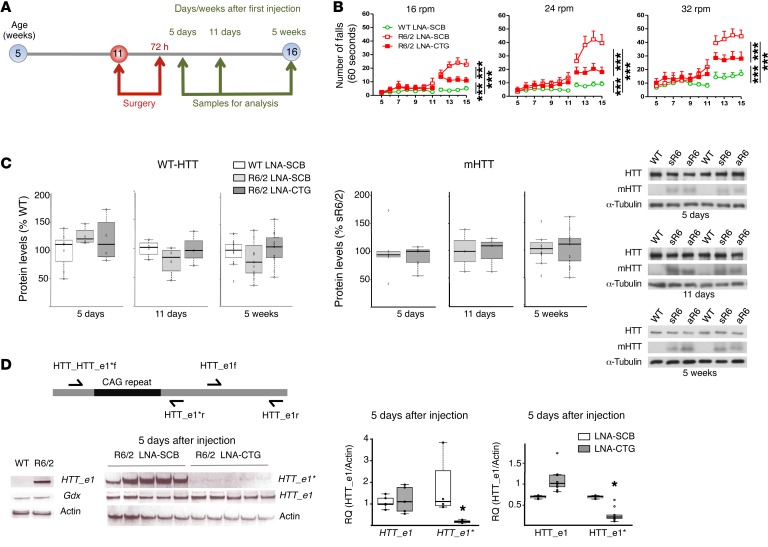
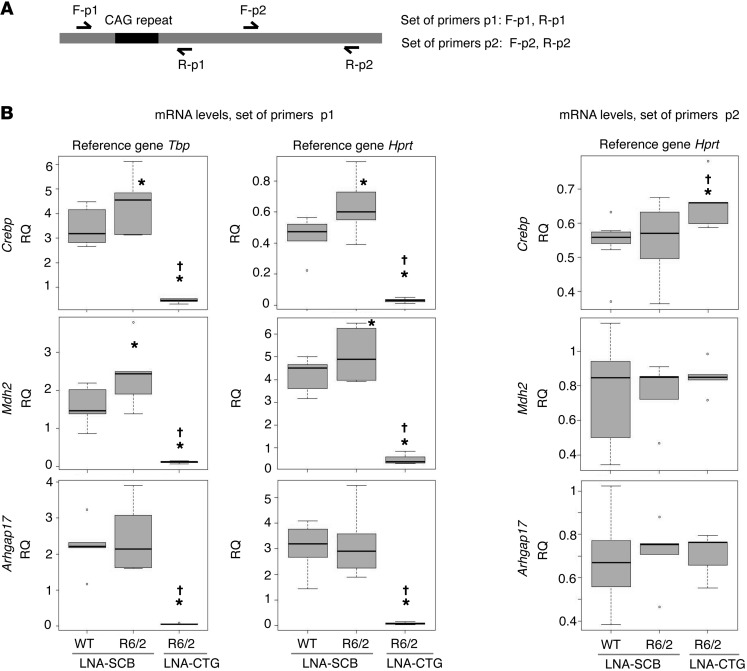
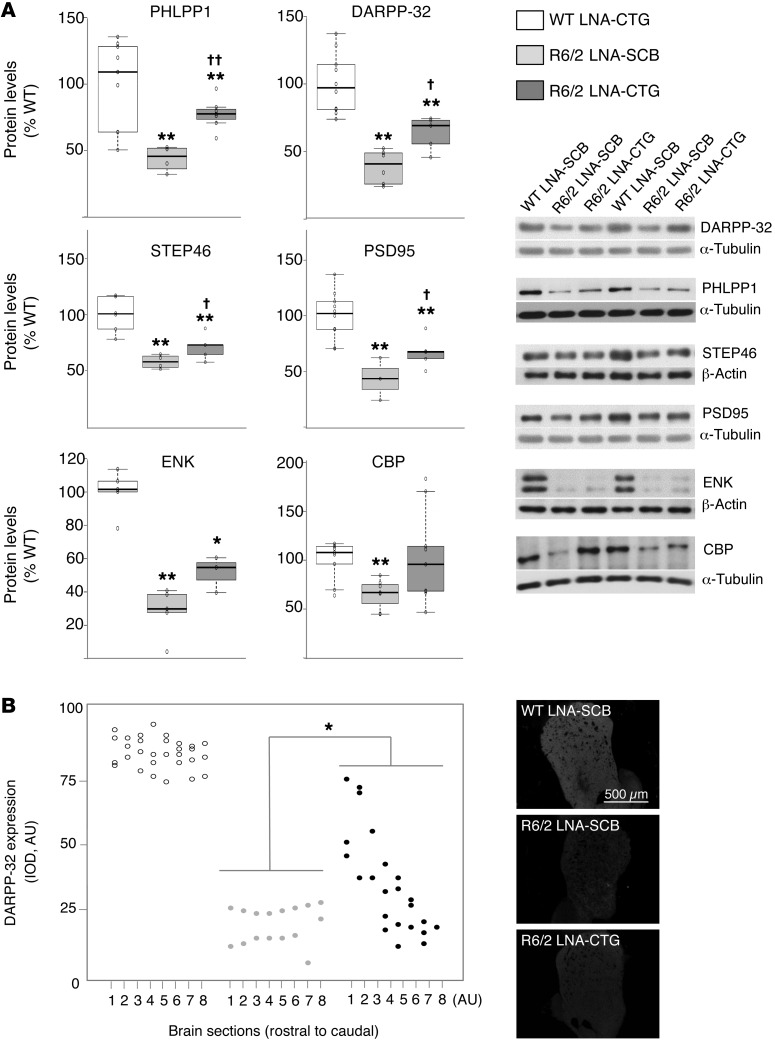
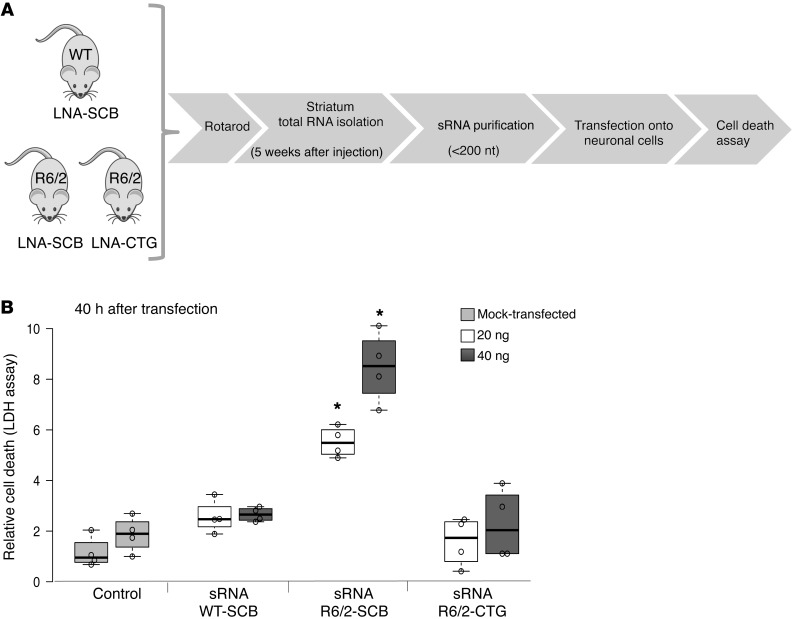
Huntington's disease (HD) is a polyglutamine disorder caused by a CAG expansion in the Huntingtin (HTT) gene exon 1. This expansion encodes a mutant protein whose abnormal function is traditionally associated with HD pathogenesis; however, recent evidence has also linked HD pathogenesis to RNA stable hairpins formed by the mutant HTT expansion. Here, we have shown that a locked nucleic acid-modified antisense oligonucleotide complementary to the CAG repeat (LNA-CTG) preferentially binds to mutant HTT without affecting HTT mRNA or protein levels. LNA-CTGs produced rapid and sustained improvement of motor deficits in an R6/2 mouse HD model that was paralleled by persistent binding of LNA-CTG to the expanded HTT exon 1 transgene. Motor improvement was accompanied by a pronounced recovery in the levels of several striatal neuronal markers severely impaired in R6/2 mice. Furthermore, in R6/2 mice, LNA-CTG blocked several pathogenic mechanisms caused by expanded CAG RNA, including small RNA toxicity and decreased Rn45s expression levels. These results suggest that LNA-CTGs promote neuroprotection by blocking the detrimental activity of CAG repeats within HTT mRNA. The present data emphasize the relevance of expanded CAG RNA to HD pathogenesis, indicate that inhibition of HTT expression is not required to reverse motor deficits, and further suggest a therapeutic potential for LNA-CTG in polyglutamine disorders.
Figures
Similar articles
-
The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain.PLoS One. 2017 Feb 9;12(2):e0171127. doi: 10.1371/journal.pone.0171127. eCollection 2017. PLoS One. 2017. PMID: 28182673 Free PMC article.
-
A pathogenic mechanism in Huntington's disease involves small CAG-repeated RNAs with neurotoxic activity.PLoS Genet. 2012;8(2):e1002481. doi: 10.1371/journal.pgen.1002481. Epub 2012 Feb 23. PLoS Genet. 2012. PMID: 22383888 Free PMC article.
-
Partial resistance to malonate-induced striatal cell death in transgenic mouse models of Huntington's disease is dependent on age and CAG repeat length.J Neurochem. 2001 Aug;78(4):694-703. doi: 10.1046/j.1471-4159.2001.00482.x. J Neurochem. 2001. PMID: 11520890
-
Multiple clinical features of Huntington's disease correlate with mutant HTT gene CAG repeat lengths and neurodegeneration.J Neurol. 2019 Mar;266(3):551-564. doi: 10.1007/s00415-018-8940-6. Epub 2018 Jun 28. J Neurol. 2019. PMID: 29956026 Review.
-
RNA toxicity induced by expanded CAG repeats in Huntington's disease.Brain Pathol. 2016 Nov;26(6):779-786. doi: 10.1111/bpa.12427. Brain Pathol. 2016. PMID: 27529325 Free PMC article. Review.
Cited by
-
Recent Advances in the Treatment of Huntington's Disease: Targeting DNA and RNA.CNS Drugs. 2020 Mar;34(3):219-228. doi: 10.1007/s40263-019-00695-3. CNS Drugs. 2020. PMID: 31933283 Review.
-
Exploring the Potential Therapeutic Approach Using Ginsenosides for the Management of Neurodegenerative Disorders.Mol Biotechnol. 2024 Jul;66(7):1520-1536. doi: 10.1007/s12033-023-00783-2. Epub 2023 Jun 18. Mol Biotechnol. 2024. PMID: 37330923 Review.
-
Some Molecular and Cellular Stress Mechanisms Associated with Neurodegenerative Diseases and Atherosclerosis.Int J Mol Sci. 2021 Jan 12;22(2):699. doi: 10.3390/ijms22020699. Int J Mol Sci. 2021. PMID: 33445687 Free PMC article. Review.
-
Protein Misfolding and Aggregation as a Therapeutic Target for Polyglutamine Diseases.Brain Sci. 2017 Oct 11;7(10):128. doi: 10.3390/brainsci7100128. Brain Sci. 2017. PMID: 29019918 Free PMC article. Review.
-
Interferon mediated neuroinflammation in polyglutamine disease is not caused by RNA toxicity.Cell Death Dis. 2020 Jan 2;11(1):3. doi: 10.1038/s41419-019-2193-x. Cell Death Dis. 2020. PMID: 31919387 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
