

Alternative Splicing May Not Be the Key to Proteome Complexity
- PMID: 27712956
- PMCID: PMC6526280
- DOI: 10.1016/j.tibs.2016.08.008
Alternative Splicing May Not Be the Key to Proteome Complexity
Abstract
Alternative splicing is commonly believed to be a major source of cellular protein diversity. However, although many thousands of alternatively spliced transcripts are routinely detected in RNA-seq studies, reliable large-scale mass spectrometry-based proteomics analyses identify only a small fraction of annotated alternative isoforms. The clearest finding from proteomics experiments is that most human genes have a single main protein isoform, while those alternative isoforms that are identified tend to be the most biologically plausible: those with the most cross-species conservation and those that do not compromise functional domains. Indeed, most alternative exons do not seem to be under selective pressure, suggesting that a large majority of predicted alternative transcripts may not even be translated into proteins.
Keywords: RNA-seq; alternative splicing; dominant isoforms; functional isoforms; homology; proteomics.
Copyright © 2016 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
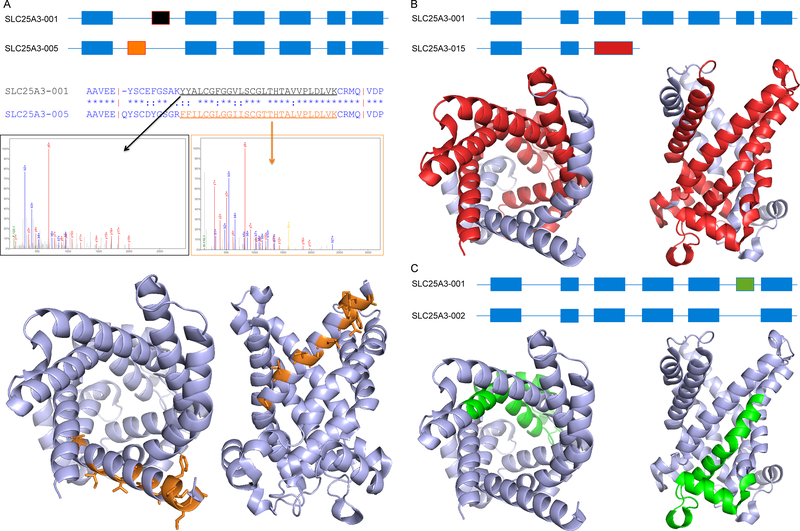
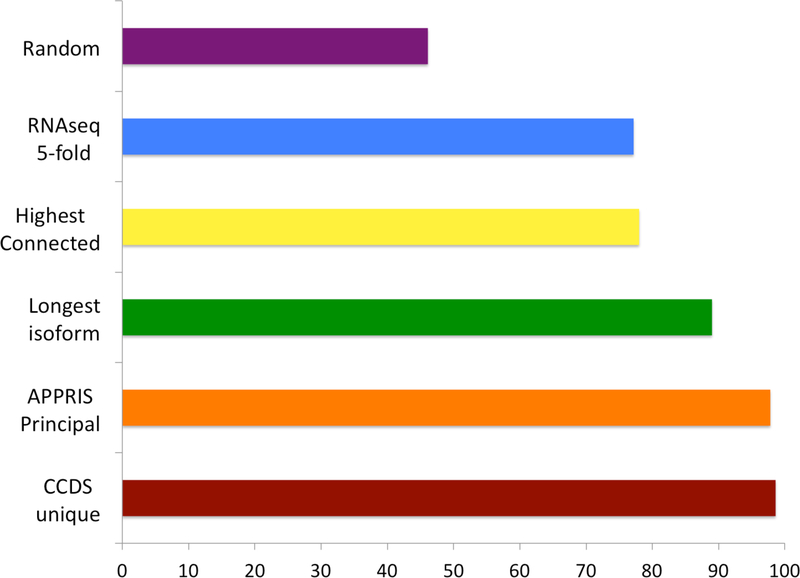
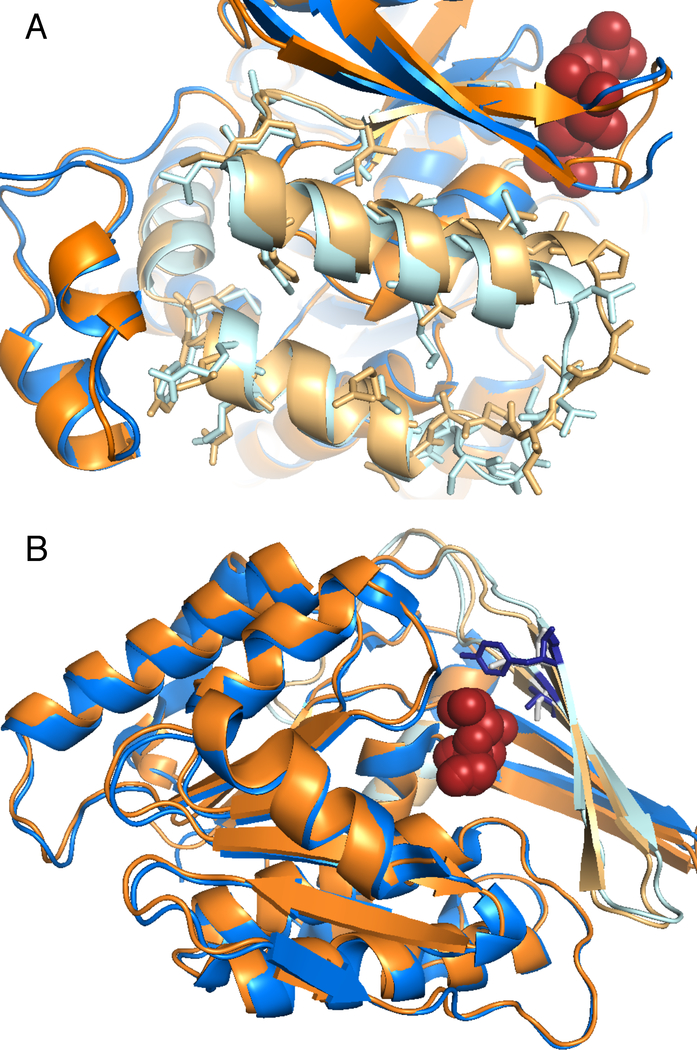
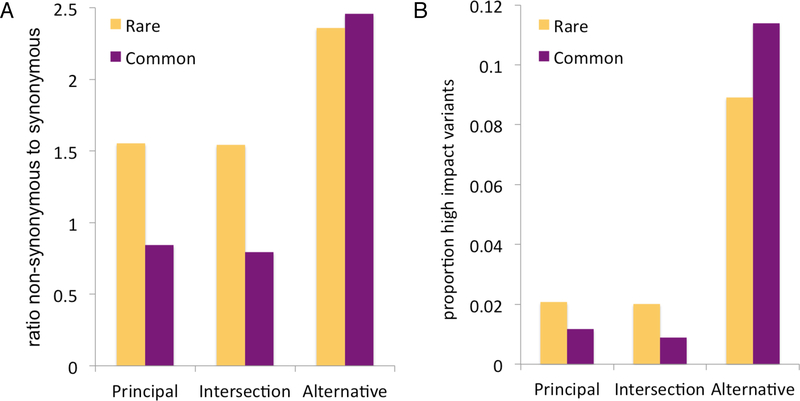
Comment in
-
The Relationship between Alternative Splicing and Proteomic Complexity.Trends Biochem Sci. 2017 Jun;42(6):407-408. doi: 10.1016/j.tibs.2017.04.001. Epub 2017 May 5. Trends Biochem Sci. 2017. PMID: 28483376 No abstract available.
-
Most Alternative Isoforms Are Not Functionally Important.Trends Biochem Sci. 2017 Jun;42(6):408-410. doi: 10.1016/j.tibs.2017.04.002. Epub 2017 May 5. Trends Biochem Sci. 2017. PMID: 28483377 Free PMC article. No abstract available.
Similar articles
-
Alternatively Spliced Homologous Exons Have Ancient Origins and Are Highly Expressed at the Protein Level.PLoS Comput Biol. 2015 Jun 10;11(6):e1004325. doi: 10.1371/journal.pcbi.1004325. eCollection 2015 Jun. PLoS Comput Biol. 2015. PMID: 26061177 Free PMC article.
-
Identification of novel alternative splicing biomarkers for breast cancer with LC/MS/MS and RNA-Seq.BMC Bioinformatics. 2020 Dec 3;21(Suppl 9):541. doi: 10.1186/s12859-020-03824-8. BMC Bioinformatics. 2020. PMID: 33272210 Free PMC article.
-
Splice-Junction-Based Mapping of Alternative Isoforms in the Human Proteome.Cell Rep. 2019 Dec 10;29(11):3751-3765.e5. doi: 10.1016/j.celrep.2019.11.026. Cell Rep. 2019. PMID: 31825849 Free PMC article.
-
Identification of Splice Variants and Isoforms in Transcriptomics and Proteomics.Annu Rev Biomed Data Sci. 2023 Aug 10;6:357-376. doi: 10.1146/annurev-biodatasci-020722-044021. Annu Rev Biomed Data Sci. 2023. PMID: 37561601 Free PMC article. Review.
-
Alternative intronic promoters in development and disease.Protoplasma. 2017 May;254(3):1201-1206. doi: 10.1007/s00709-016-1071-y. Epub 2017 Jan 11. Protoplasma. 2017. PMID: 28078440 Review.
Cited by
-
Alternative Splicing: A New Therapeutic Target for Ovarian Cancer.Technol Cancer Res Treat. 2022 Jan-Dec;21:15330338211067911. doi: 10.1177/15330338211067911. Technol Cancer Res Treat. 2022. PMID: 35343831 Free PMC article.
-
Full-length transcriptome reconstruction reveals a large diversity of RNA and protein isoforms in rat hippocampus.Nat Commun. 2019 Nov 1;10(1):5009. doi: 10.1038/s41467-019-13037-0. Nat Commun. 2019. PMID: 31676752 Free PMC article.
-
The Common Partner of Several Methyltransferases TRMT112 Regulates the Expression of N6AMT1 Isoforms in Mammalian Cells.Biomolecules. 2019 Aug 28;9(9):422. doi: 10.3390/biom9090422. Biomolecules. 2019. PMID: 31466382 Free PMC article.
-
Modeling the structural implications of an alternatively spliced Exoc3l2, a paralog of the tunneling nanotube-forming M-Sec.PLoS One. 2018 Aug 7;13(8):e0201557. doi: 10.1371/journal.pone.0201557. eCollection 2018. PLoS One. 2018. PMID: 30086153 Free PMC article.
-
Interplays between cis- and trans-Acting Factors for Alternative Splicing in Response to Environmental Changes during Biological Invasions of Ascidians.Int J Mol Sci. 2023 Oct 5;24(19):14921. doi: 10.3390/ijms241914921. Int J Mol Sci. 2023. PMID: 37834365 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
