

Decreased somatic hypermutation induces an impaired peripheral B cell tolerance checkpoint
- PMID: 27701145
- PMCID: PMC5096912
- DOI: 10.1172/JCI84645
Decreased somatic hypermutation induces an impaired peripheral B cell tolerance checkpoint
Abstract
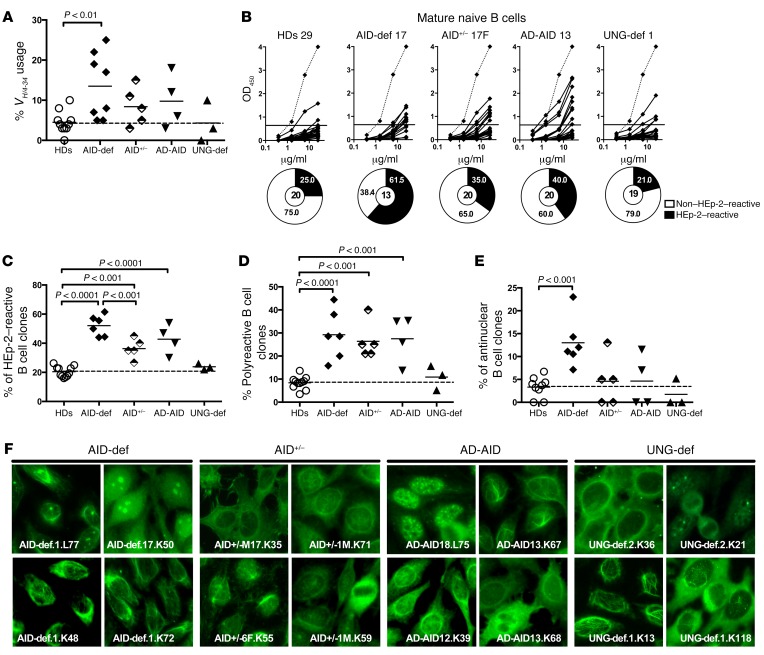
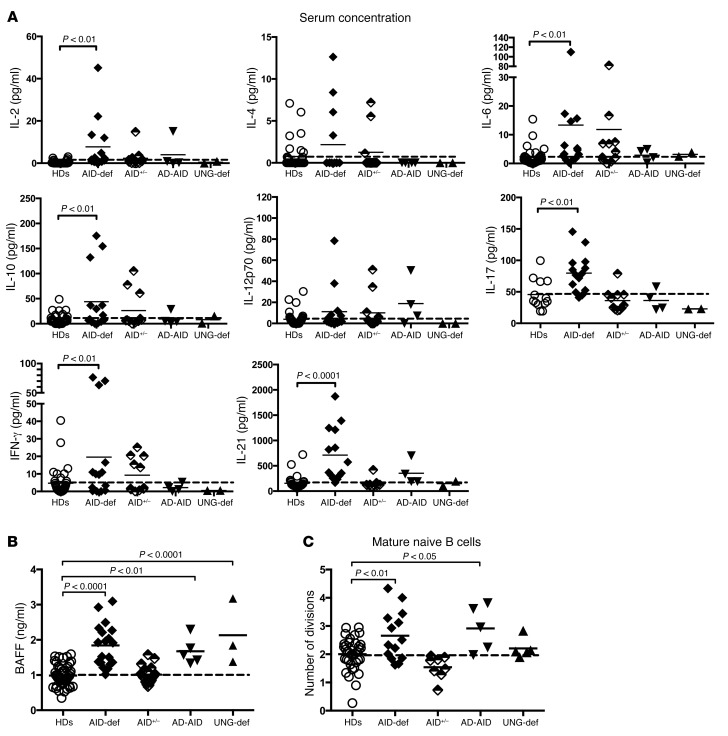
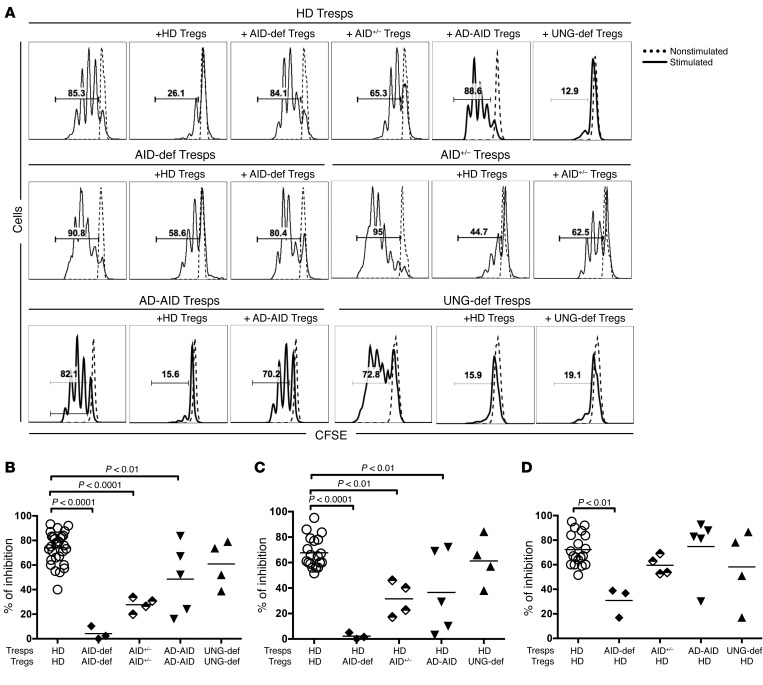
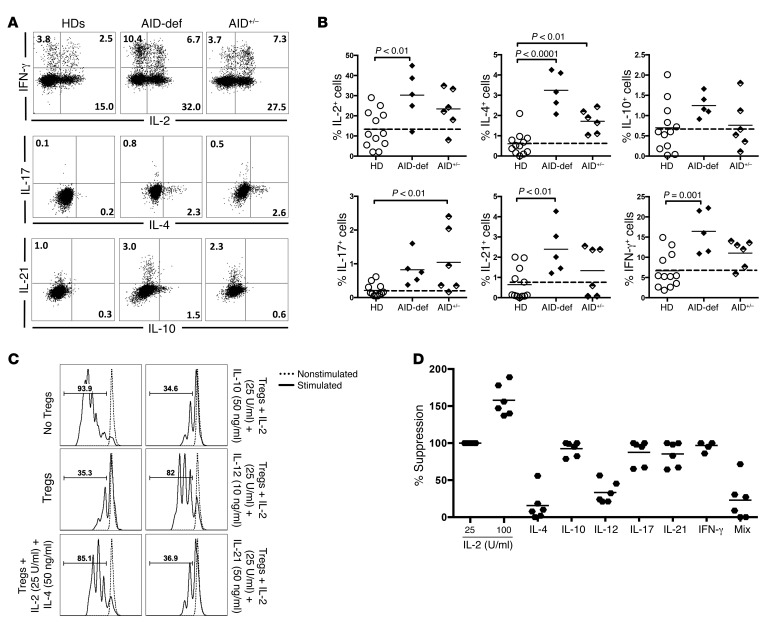
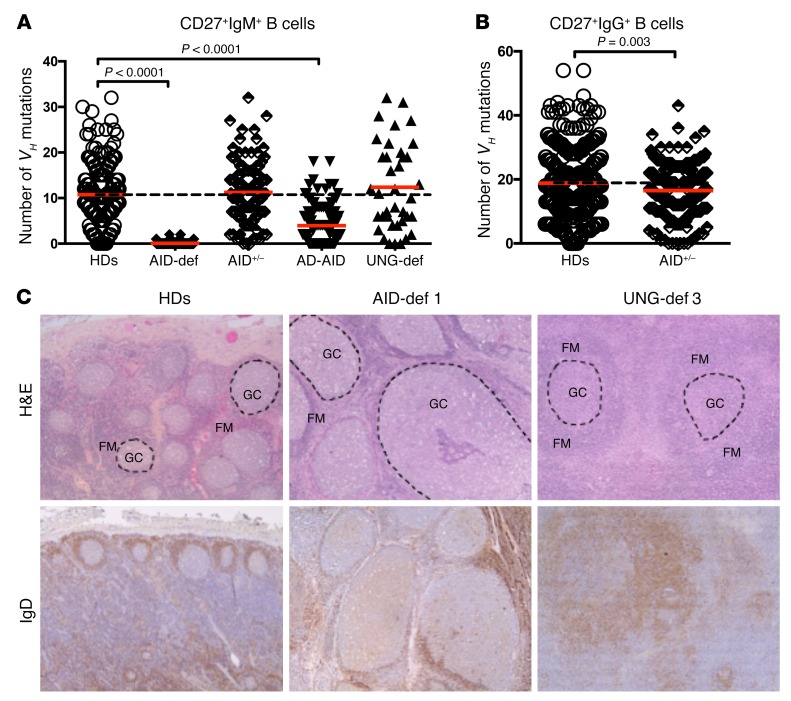
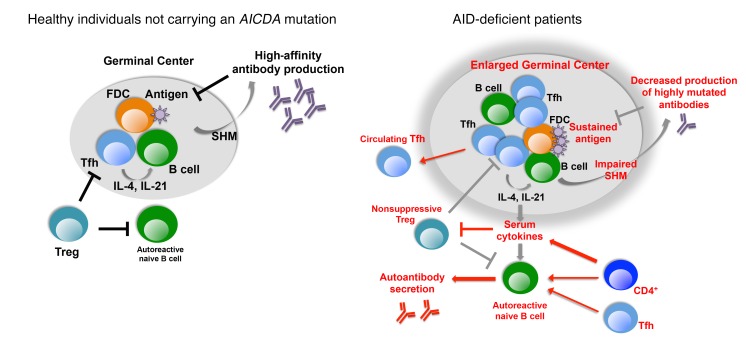
Patients with mutations in AICDA, which encodes activation-induced cytidine deaminase (AID), display an impaired peripheral B cell tolerance. AID mediates class-switch recombination (CSR) and somatic hypermutation (SHM) in B cells, but the mechanism by which AID prevents the accumulation of autoreactive B cells in blood is unclear. Here, we analyzed B cell tolerance in AID-deficient patients, patients with autosomal dominant AID mutations (AD-AID), asymptomatic AICDA heterozygotes (AID+/-), and patients with uracil N-glycosylase (UNG) deficiency, which impairs CSR but not SHM. The low frequency of autoreactive mature naive B cells in UNG-deficient patients resembled that of healthy subjects, revealing that impaired CSR does not interfere with the peripheral B cell tolerance checkpoint. In contrast, we observed decreased frequencies of SHM in memory B cells from AD-AID patients and AID+/- subjects, who were unable to prevent the accumulation of autoreactive mature naive B cells. In addition, the individuals with AICDA mutations, but not UNG-deficient patients, displayed Tregs with defective suppressive capacity that correlated with increases in circulating T follicular helper cells and enhanced cytokine production. We conclude that SHM, but not CSR, regulates peripheral B cell tolerance through the production of mutated antibodies that clear antigens and prevent sustained interleukin secretions that interfere with Treg function.
Figures


Similar articles
-
Analysis of somatic hypermutations in the IgM switch region in human B cells.J Allergy Clin Immunol. 2014 Aug;134(2):411-9. doi: 10.1016/j.jaci.2014.02.043. Epub 2014 May 15. J Allergy Clin Immunol. 2014. PMID: 24836470
-
Pathophysiology of B-cell intrinsic immunoglobulin class switch recombination deficiencies.Adv Immunol. 2007;94:275-306. doi: 10.1016/S0065-2776(06)94009-7. Adv Immunol. 2007. PMID: 17560278 Review.
-
A Novel AICDA Splice-Site Mutation in Two Siblings with HIGM2 Permits Somatic Hypermutation but Abrogates Mutational Targeting.J Clin Immunol. 2022 May;42(4):771-782. doi: 10.1007/s10875-022-01233-5. Epub 2022 Mar 5. J Clin Immunol. 2022. PMID: 35246784 Free PMC article.
-
B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil.J Exp Med. 2005 Jun 20;201(12):2011-21. doi: 10.1084/jem.20050042. J Exp Med. 2005. PMID: 15967827 Free PMC article.
-
Opinion: uracil DNA glycosylase (UNG) plays distinct and non-canonical roles in somatic hypermutation and class switch recombination.Int Immunol. 2014 Oct;26(10):575-8. doi: 10.1093/intimm/dxu071. Epub 2014 Jul 3. Int Immunol. 2014. PMID: 24994819 Free PMC article. Review.
Cited by
-
Infant Antibody Repertoires during the First Two Years of Influenza Vaccination.mBio. 2022 Dec 20;13(6):e0254622. doi: 10.1128/mbio.02546-22. Epub 2022 Oct 31. mBio. 2022. PMID: 36314798 Free PMC article.
-
Defective Early B Cell Tolerance Checkpoints in Patients With Systemic Sclerosis Allow the Production of Self Antigen-Specific Clones.Arthritis Rheumatol. 2022 Feb;74(2):307-317. doi: 10.1002/art.41927. Epub 2021 Dec 16. Arthritis Rheumatol. 2022. PMID: 34279059 Free PMC article.
-
Autoantibody Profiling in Plasma of Dengue Virus-Infected Individuals.Pathogens. 2020 Dec 18;9(12):1060. doi: 10.3390/pathogens9121060. Pathogens. 2020. PMID: 33352902 Free PMC article.
-
Balancing functions of regulatory T cells in mosquito-borne viral infections.Emerg Microbes Infect. 2024 Dec;13(1):2304061. doi: 10.1080/22221751.2024.2304061. Epub 2024 Jan 25. Emerg Microbes Infect. 2024. PMID: 38192073 Free PMC article. Review.
-
Minding the gap: The impact of B-cell tolerance on the microbial antibody repertoire.Immunol Rev. 2019 Nov;292(1):24-36. doi: 10.1111/imr.12805. Epub 2019 Sep 27. Immunol Rev. 2019. PMID: 31559648 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
