

Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways
- PMID: 27630174
- PMCID: PMC5243224
- DOI: 10.1152/physrev.00004.2016
Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways
Abstract
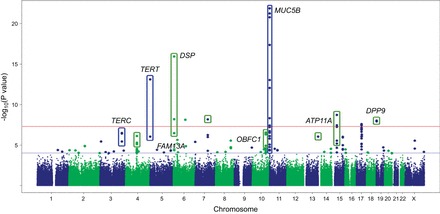
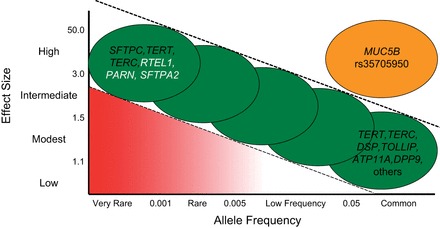
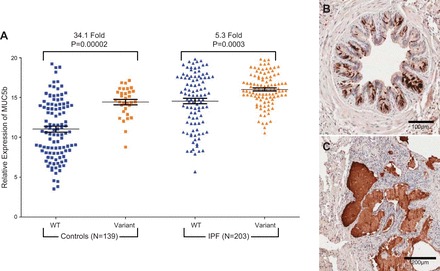
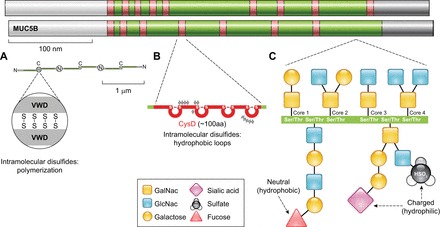
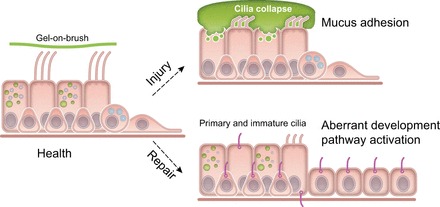
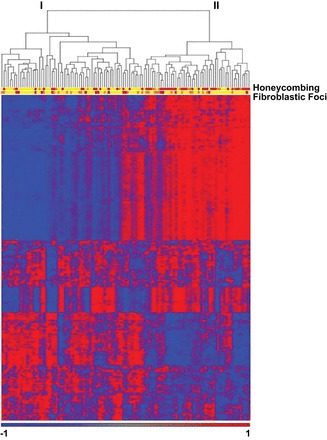
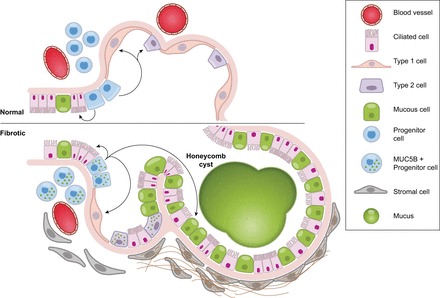
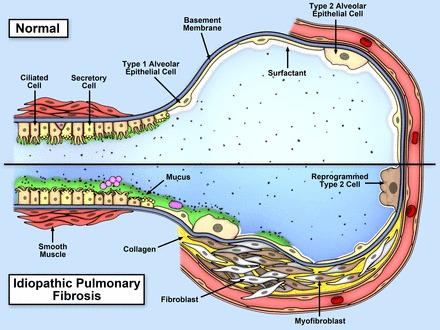
Idiopathic pulmonary fibrosis (IPF) is an incurable complex genetic disorder that is associated with sequence changes in 7 genes (MUC5B, TERT, TERC, RTEL1, PARN, SFTPC, and SFTPA2) and with variants in at least 11 novel loci. We have previously found that 1) a common gain-of-function promoter variant in MUC5B rs35705950 is the strongest risk factor (genetic and otherwise), accounting for 30-35% of the risk of developing IPF, a disease that was previously considered idiopathic; 2) the MUC5B promoter variant can potentially be used to identify individuals with preclinical pulmonary fibrosis and is predictive of radiologic progression of preclinical pulmonary fibrosis; and 3) MUC5B may be involved in the pathogenesis of pulmonary fibrosis with MUC5B message and protein expressed in bronchiolo-alveolar epithelia of IPF and the characteristic IPF honeycomb cysts. Based on these considerations, we hypothesize that excessive production of MUC5B either enhances injury due to reduced mucociliary clearance or impedes repair consequent to disruption of normal regenerative mechanisms in the distal lung. In aggregate, these novel considerations should have broad impact, resulting in specific etiologic targets, early detection of disease, and novel biologic pathways for use in the design of future intervention, prevention, and mechanistic studies of IPF.
Copyright © 2016 the American Physiological Society.
Figures




Similar articles
-
Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice.Nat Commun. 2018 Dec 18;9(1):5363. doi: 10.1038/s41467-018-07768-9. Nat Commun. 2018. PMID: 30560893 Free PMC article.
-
Idiopathic Pulmonary Fibrosis Is a Genetic Disease Involving Mucus and the Peripheral Airways.Ann Am Thorac Soc. 2018 Nov;15(Suppl 3):S192-S197. doi: 10.1513/AnnalsATS.201802-144AW. Ann Am Thorac Soc. 2018. PMID: 30431344 Free PMC article. Review.
-
MUC5B and Idiopathic Pulmonary Fibrosis.Ann Am Thorac Soc. 2015 Nov;12 Suppl 2(Suppl 2):S193-9. doi: 10.1513/AnnalsATS.201503-110AW. Ann Am Thorac Soc. 2015. PMID: 26595739 Free PMC article.
-
Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: a candidate gene sequencing study.Lancet Respir Med. 2018 Aug;6(8):603-614. doi: 10.1016/S2213-2600(18)30135-8. Epub 2018 Jun 18. Lancet Respir Med. 2018. PMID: 29891356 Free PMC article.
-
The Possible Pathogenesis of Idiopathic Pulmonary Fibrosis considering MUC5B.Biomed Res Int. 2019 Jun 11;2019:9712464. doi: 10.1155/2019/9712464. eCollection 2019. Biomed Res Int. 2019. PMID: 31309122 Free PMC article. Review.
Cited by
-
The relationship between MUC5B promoter, TERT polymorphisms and telomere lengths with radiographic extent and survival in a Chinese IPF cohort.Sci Rep. 2019 Oct 25;9(1):15307. doi: 10.1038/s41598-019-51902-6. Sci Rep. 2019. PMID: 31653936 Free PMC article.
-
The role of antifibrotics in the treatment of rheumatoid arthritis-associated interstitial lung disease.Ther Adv Musculoskelet Dis. 2022 Feb 15;14:1759720X221074457. doi: 10.1177/1759720X221074457. eCollection 2022. Ther Adv Musculoskelet Dis. 2022. PMID: 35186127 Free PMC article. Review.
-
The Association Between MUC5B Mutations and Clinical Outcome in Patients with Rheumatoid Arthritis-Associated Interstitial Lung Disease: A Retrospective Exploratory Study in China.Med Sci Monit. 2020 Mar 6;26:e920137. doi: 10.12659/MSM.920137. Med Sci Monit. 2020. PMID: 32142504 Free PMC article.
-
Air Pollution Exposure and Interstitial Lung Features in SPIROMICS Participants with Chronic Obstructive Pulmonary Disease.Ann Am Thorac Soc. 2024 Sep;21(9):1251-1260. doi: 10.1513/AnnalsATS.202308-741OC. Ann Am Thorac Soc. 2024. PMID: 38568439
-
Recent advances in the treatment of systemic sclerosis associated interstitial lung disease.Front Med (Lausanne). 2023 Mar 24;10:1155771. doi: 10.3389/fmed.2023.1155771. eCollection 2023. Front Med (Lausanne). 2023. PMID: 37035331 Free PMC article. Review.
References
-
- Antonarakis SE, Chakravarti A, Cohen JC, Hardy J. Mendelian disorders and multifactorial traits: the big divide or one for all? Nat Rev Genet 11: 380–384, 2010. - PubMed
-
- Araki T, Putman RK, Hatabu H, Gao W, Dupuis J, Latourelle JC, Nishino M, Zazueta OE, Kurugol S, Ross JC, Estepar RS, Schwartz DA, Rosas IO, Washko GR, O'Connor GT, Hunninghake GM. Development and progression of interstitial lung abnormalities in the Framingham Heart Study. Am J Respir Crit Care Med. In press. - PMC - PubMed
-
- Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 356: 1317–1326, 2007. - PubMed
-
- Baek HA, Kim do S, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol 46: 731–739, 2012. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 HL092870/HL/NHLBI NIH HHS/United States
- R21 ES023384/ES/NIEHS NIH HHS/United States
- R21 HL120770/HL/NHLBI NIH HHS/United States
- T32 GM008497/GM/NIGMS NIH HHS/United States
- R01 HL080396/HL/NHLBI NIH HHS/United States
- I01 BX001534/BX/BLRD VA/United States
- R01 HL097163/HL/NHLBI NIH HHS/United States
- R25 ES025476/ES/NIEHS NIH HHS/United States
- R01 HL130938/HL/NHLBI NIH HHS/United States
- R33 HL120770/HL/NHLBI NIH HHS/United States
- UH2 HL123442/HL/NHLBI NIH HHS/United States
- UH3 HL123442/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources