

A computational approach to map nucleosome positions and alternative chromatin states with base pair resolution
- PMID: 27623011
- PMCID: PMC5094857
- DOI: 10.7554/eLife.16970
A computational approach to map nucleosome positions and alternative chromatin states with base pair resolution
Abstract
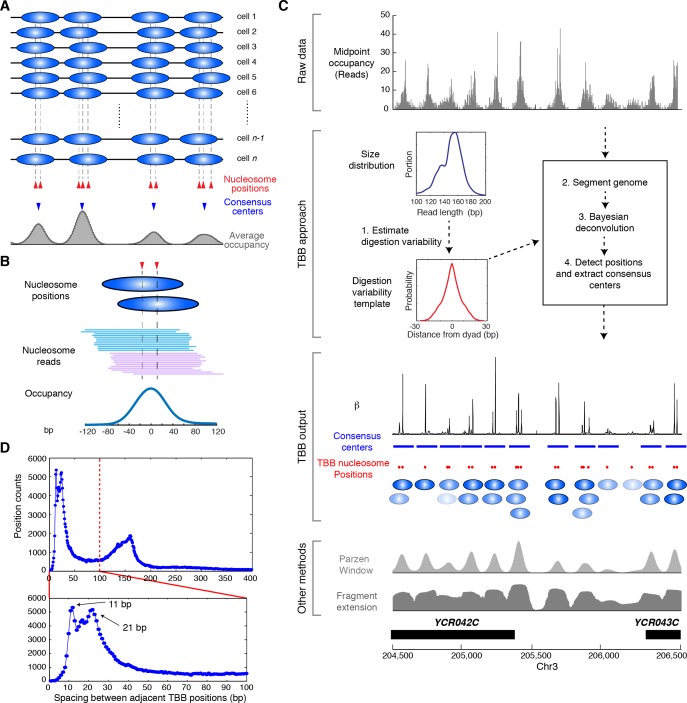
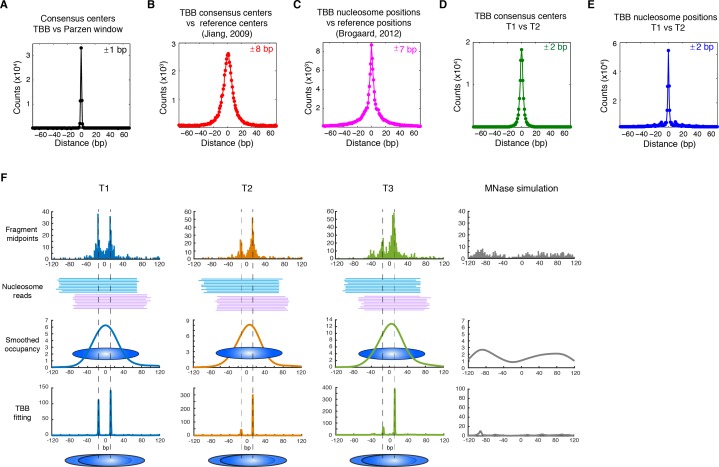
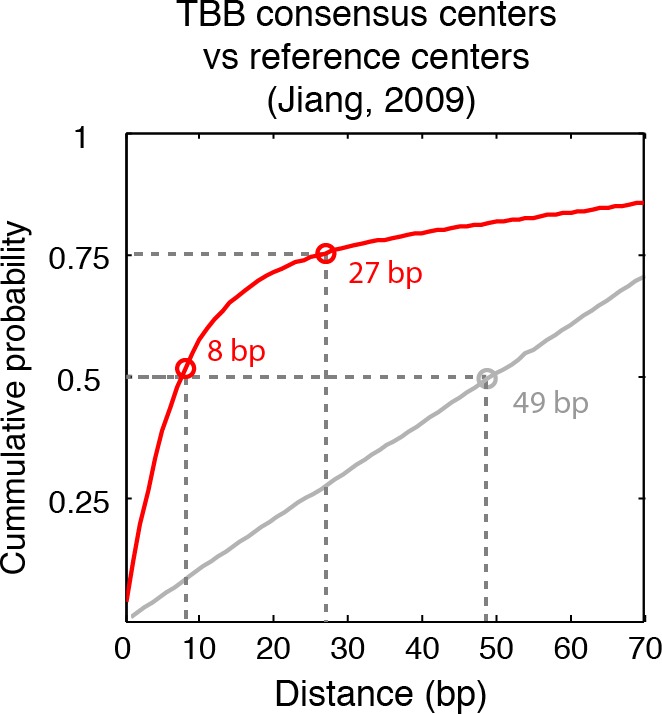
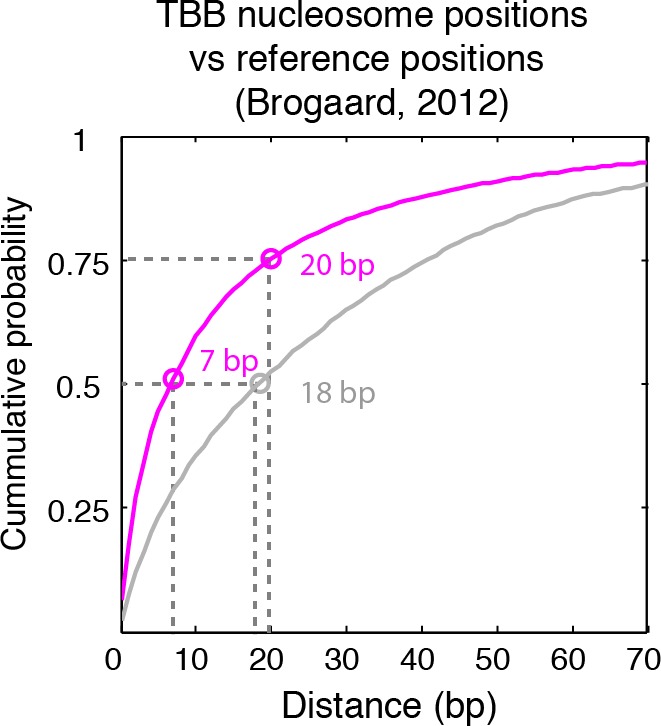
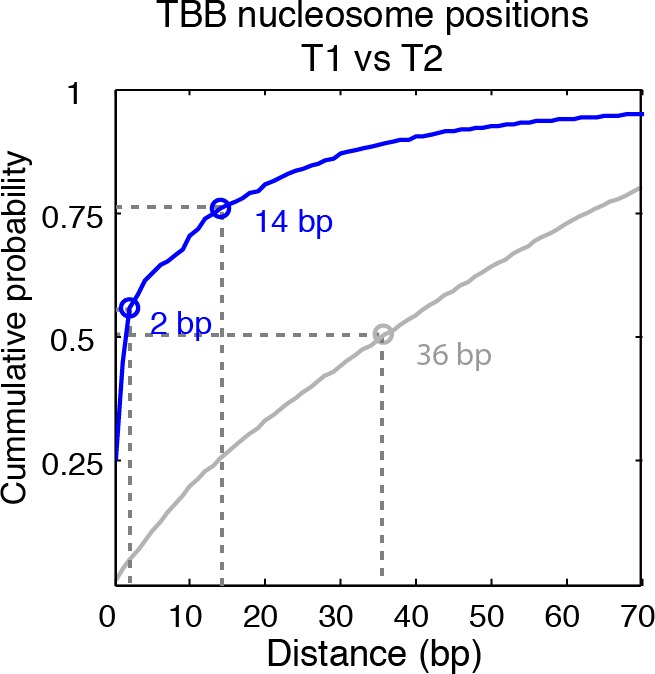
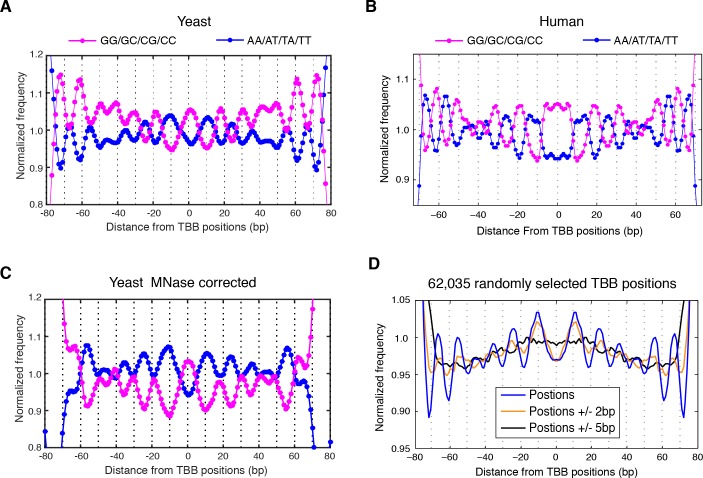
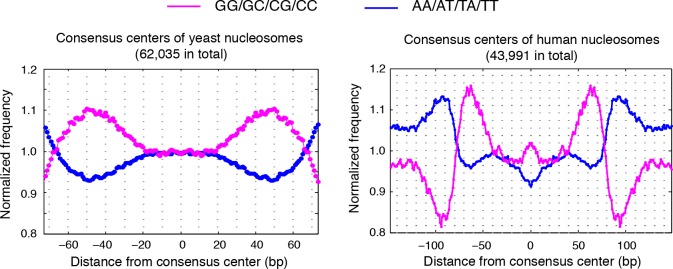
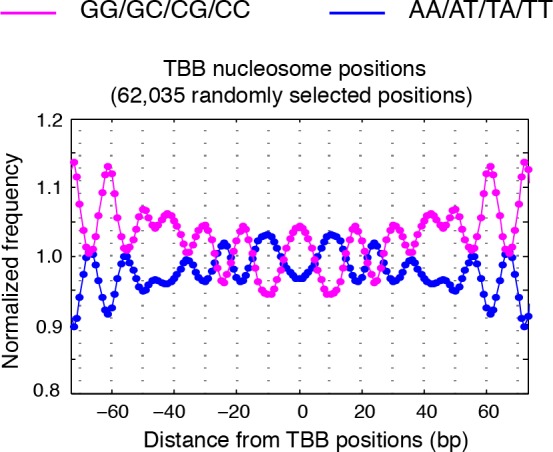
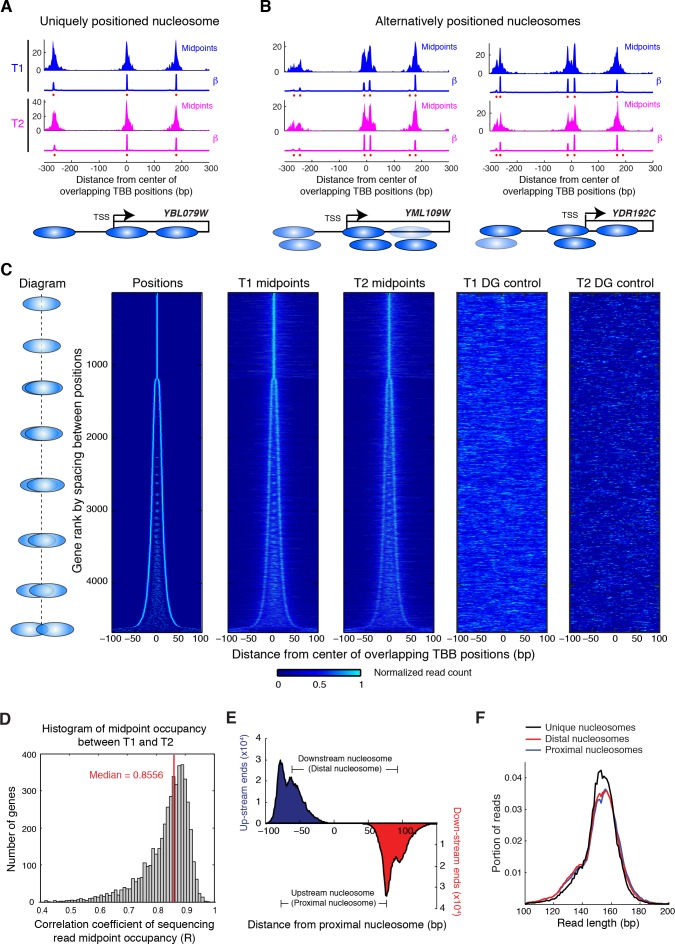
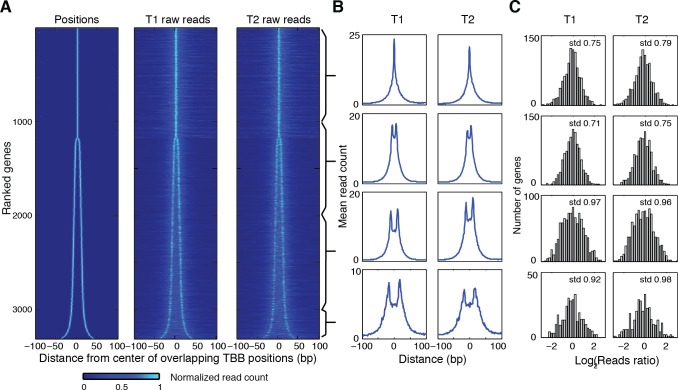
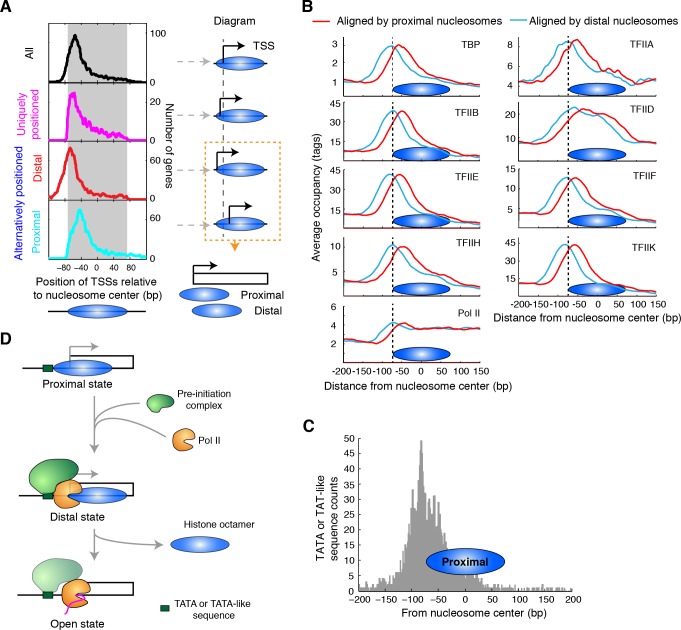
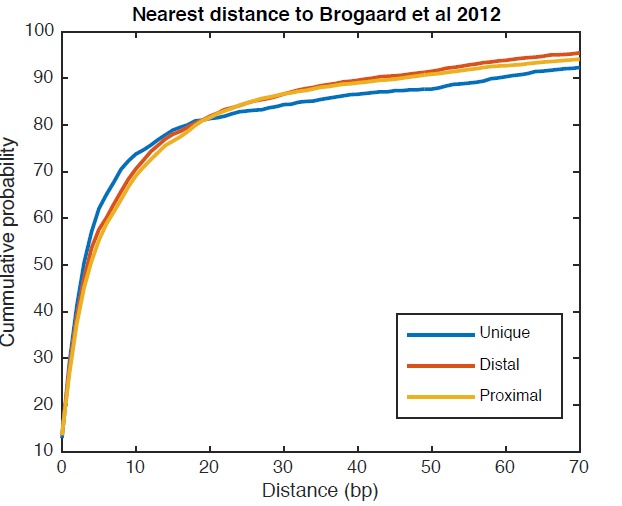
Understanding chromatin function requires knowing the precise location of nucleosomes. MNase-seq methods have been widely applied to characterize nucleosome organization in vivo, but generally lack the accuracy to determine the precise nucleosome positions. Here we develop a computational approach leveraging digestion variability to determine nucleosome positions at a base-pair resolution from MNase-seq data. We generate a variability template as a simple error model for how MNase digestion affects the mapping of individual nucleosomes. Applied to both yeast and human cells, this analysis reveals that alternatively positioned nucleosomes are prevalent and create significant heterogeneity in a cell population. We show that the periodic occurrences of dinucleotide sequences relative to nucleosome dyads can be directly determined from genome-wide nucleosome positions from MNase-seq. Alternatively positioned nucleosomes near transcription start sites likely represent different states of promoter nucleosomes during transcription initiation. Our method can be applied to map nucleosome positions in diverse organisms at base-pair resolution.
Keywords: MNase-seq; S. cerevisiae; base-pair resolution; computational biology; evolutionary biology; gene regulation; genomics; heterogenity; human; nucleosome position; systems biology; template-based model.
Conflict of interest statement
EKO: President at the Howard Hughes Medical Institute, one of the three founding funders of eLife. The other authors declare that no competing interests exist.
Figures











Similar articles
-
Subtracting the sequence bias from partially digested MNase-seq data reveals a general contribution of TFIIS to nucleosome positioning.Epigenetics Chromatin. 2017 Dec 7;10(1):58. doi: 10.1186/s13072-017-0165-x. Epigenetics Chromatin. 2017. PMID: 29212533 Free PMC article.
-
Mapping nucleosome positions using DNase-seq.Genome Res. 2016 Mar;26(3):351-64. doi: 10.1101/gr.195602.115. Epub 2016 Jan 15. Genome Res. 2016. PMID: 26772197 Free PMC article.
-
Profiling Nucleosome Occupancy by MNase-seq: Experimental Protocol and Computational Analysis.Methods Mol Biol. 2018;1675:167-181. doi: 10.1007/978-1-4939-7318-7_11. Methods Mol Biol. 2018. PMID: 29052192
-
Nucleosome positioning and spacing: from genome-wide maps to single arrays.Essays Biochem. 2019 Apr 23;63(1):5-14. doi: 10.1042/EBC20180058. Print 2019 Apr 23. Essays Biochem. 2019. PMID: 31015380 Review.
-
Nucleosome positioning and gene regulation: advances through genomics.Nat Rev Genet. 2009 Mar;10(3):161-72. doi: 10.1038/nrg2522. Nat Rev Genet. 2009. PMID: 19204718 Free PMC article. Review.
Cited by
-
SEM: sized-based expectation maximization for characterizing nucleosome positions and subtypes.bioRxiv [Preprint]. 2023 Oct 20:2023.10.17.562727. doi: 10.1101/2023.10.17.562727. bioRxiv. 2023. Update in: Genome Res. 2024 Oct 11;34(9):1334-1343. doi: 10.1101/gr.279138.124 PMID: 37904910 Free PMC article. Updated. Preprint.
-
Hi-BDiSCO: folding 3D mesoscale genome structures from Hi-C data using brownian dynamics.Nucleic Acids Res. 2024 Jan 25;52(2):583-599. doi: 10.1093/nar/gkad1121. Nucleic Acids Res. 2024. PMID: 38015443 Free PMC article.
-
Control of Inflammatory Response by Tissue Microenvironment.bioRxiv [Preprint]. 2024 May 14:2024.05.10.592432. doi: 10.1101/2024.05.10.592432. bioRxiv. 2024. PMID: 38798655 Free PMC article. Preprint.
-
Understanding nucleosome dynamics and their links to gene expression and DNA replication.Nat Rev Mol Cell Biol. 2017 Sep;18(9):548-562. doi: 10.1038/nrm.2017.47. Epub 2017 May 24. Nat Rev Mol Cell Biol. 2017. PMID: 28537572 Free PMC article. Review.
-
Chromatin accessibility profiling methods.Nat Rev Methods Primers. 2021;1:10. doi: 10.1038/s43586-020-00008-9. Epub 2021 Jan 21. Nat Rev Methods Primers. 2021. PMID: 38410680 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
