

Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson's Disease
- PMID: 27618216
- PMCID: PMC5135570
- DOI: 10.1016/j.stem.2016.08.002
Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson's Disease
Abstract
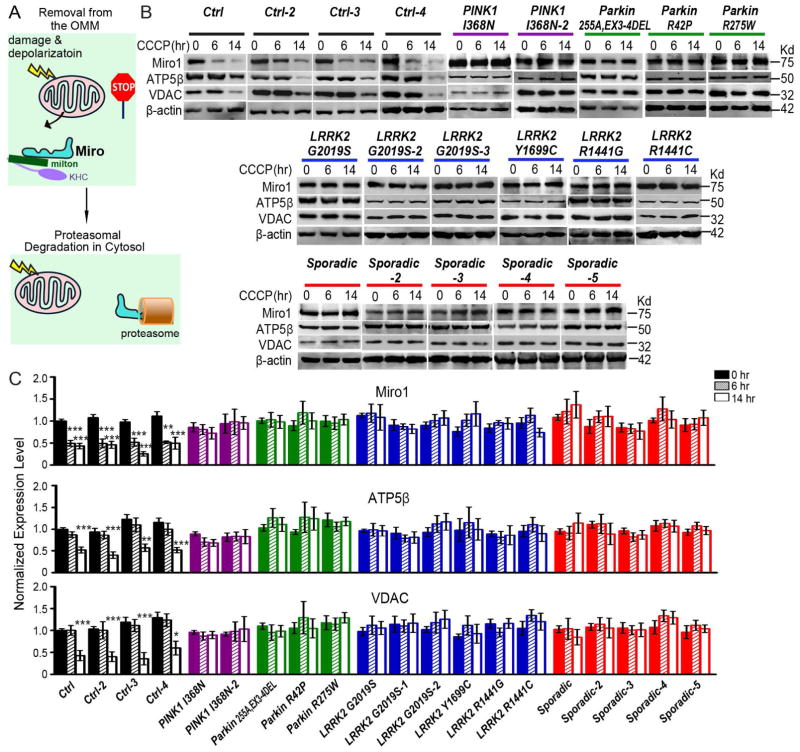
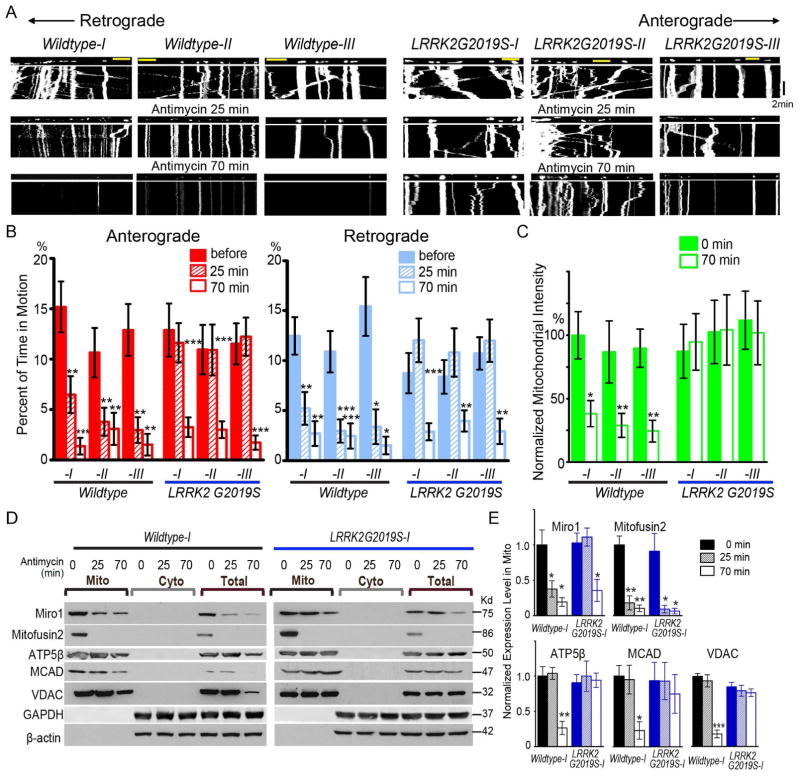
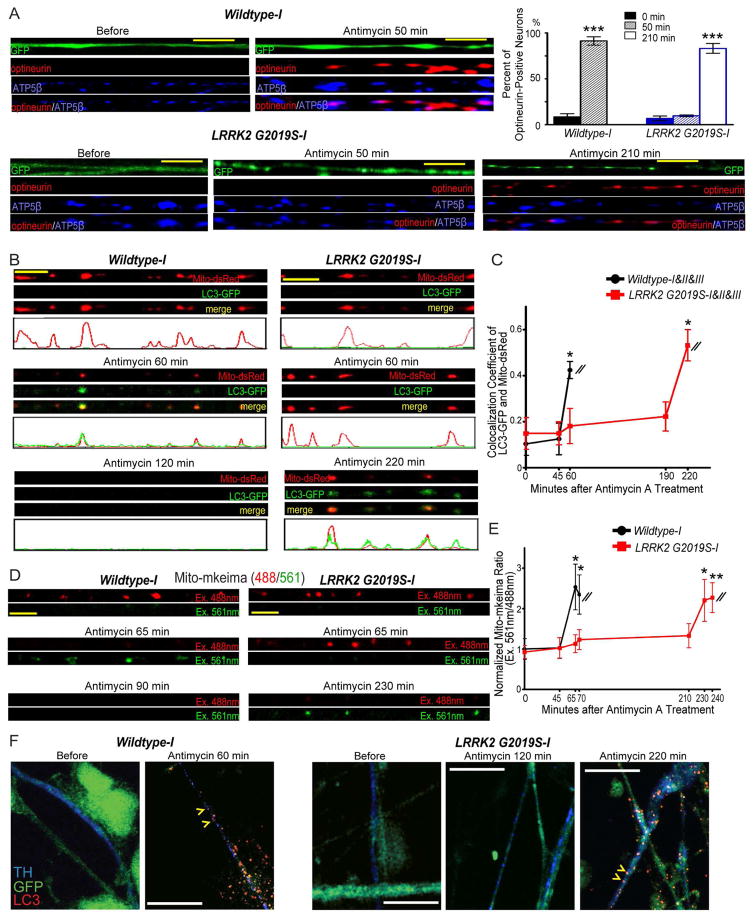
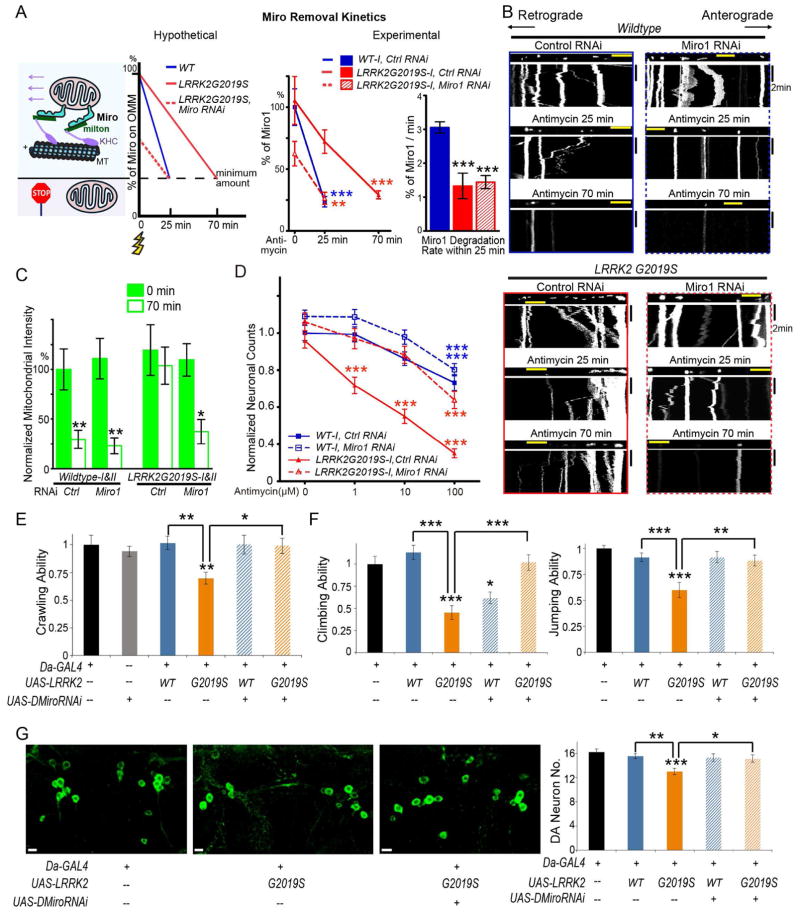
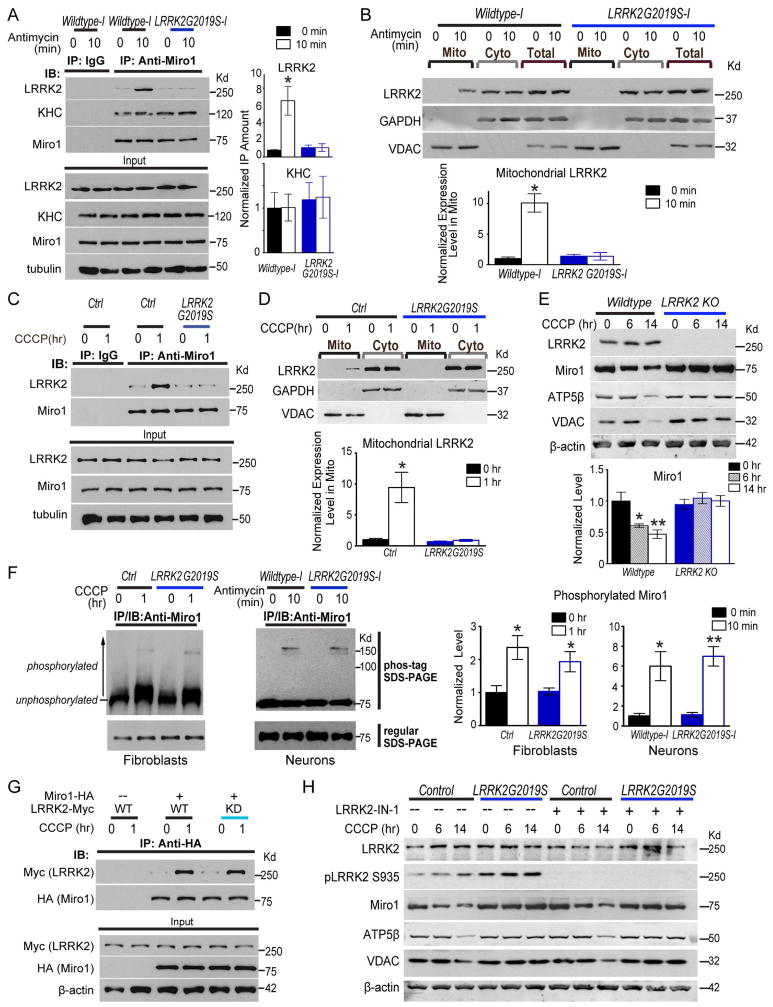
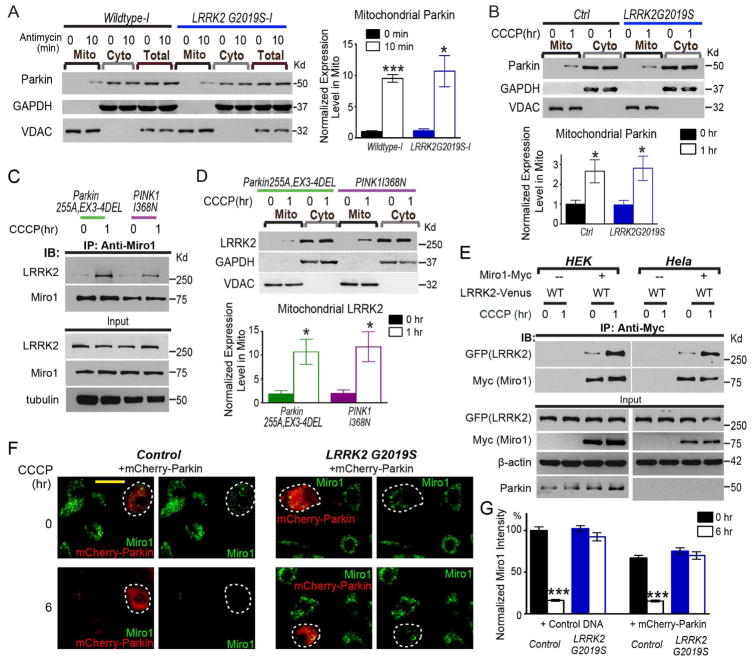
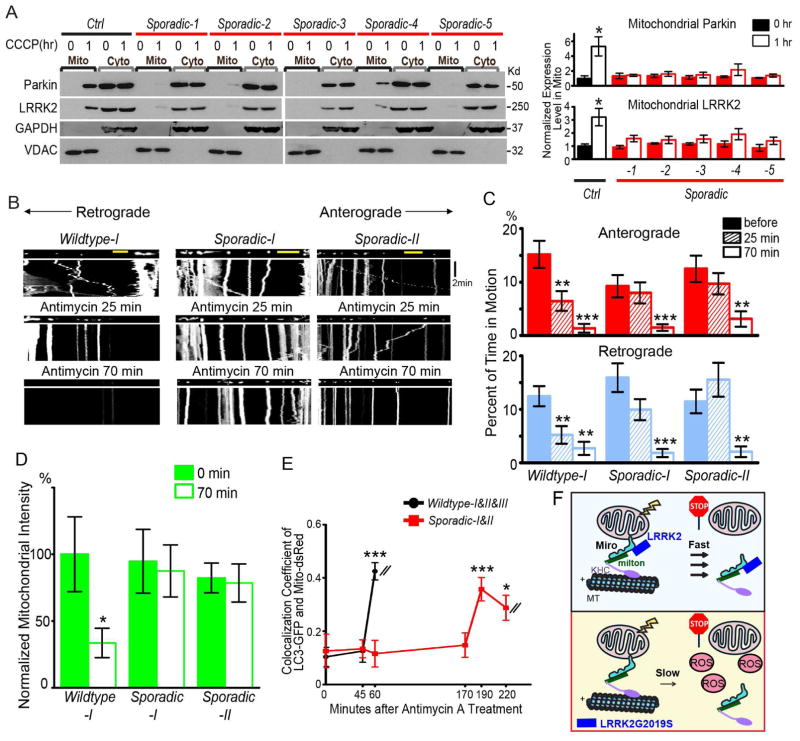
Mitochondrial movements are tightly controlled to maintain energy homeostasis and prevent oxidative stress. Miro is an outer mitochondrial membrane protein that anchors mitochondria to microtubule motors and is removed to stop mitochondrial motility as an early step in the clearance of dysfunctional mitochondria. Here, using human induced pluripotent stem cell (iPSC)-derived neurons and other complementary models, we build on a previous connection of Parkinson's disease (PD)-linked PINK1 and Parkin to Miro by showing that a third PD-related protein, LRRK2, promotes Miro removal by forming a complex with Miro. Pathogenic LRRK2G2019S disrupts this function, delaying the arrest of damaged mitochondria and consequently slowing the initiation of mitophagy. Remarkably, partial reduction of Miro levels in LRRK2G2019S human neuron and Drosophila PD models rescues neurodegeneration. Miro degradation and mitochondrial motility are also impaired in sporadic PD patients. We reveal that prolonged retention of Miro, and the downstream consequences that ensue, may constitute a central component of PD pathogenesis.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Inappropriate trafficking of damaged mitochondria in Parkinson's disease.Stem Cell Investig. 2017 Feb 27;4:17. doi: 10.21037/sci.2017.02.07. eCollection 2017. Stem Cell Investig. 2017. PMID: 28275647 Free PMC article. No abstract available.
-
"Miro" in Parkinson's disease: Here, there, everywhere!Mov Disord. 2017 Jun;32(6):839. doi: 10.1002/mds.26975. Epub 2017 May 8. Mov Disord. 2017. PMID: 28597558 No abstract available.
Similar articles
-
PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility.Cell. 2011 Nov 11;147(4):893-906. doi: 10.1016/j.cell.2011.10.018. Cell. 2011. PMID: 22078885 Free PMC article.
-
Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models.Proc Natl Acad Sci U S A. 2018 Sep 18;115(38):E8844-E8853. doi: 10.1073/pnas.1721136115. Epub 2018 Sep 5. Proc Natl Acad Sci U S A. 2018. PMID: 30185553 Free PMC article.
-
Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson's models.Acta Neuropathol. 2018 Oct;136(4):607-620. doi: 10.1007/s00401-018-1873-4. Epub 2018 Jun 9. Acta Neuropathol. 2018. PMID: 29923074 Free PMC article.
-
Mechanisms of neurodegeneration in Parkinson's disease: keep neurons in the PINK1.Mech Ageing Dev. 2020 Jul;189:111277. doi: 10.1016/j.mad.2020.111277. Epub 2020 Jun 3. Mech Ageing Dev. 2020. PMID: 32504621 Review.
-
Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson's disease.Exp Neurol. 2009 Aug;218(2):235-46. doi: 10.1016/j.expneurol.2009.03.006. Epub 2009 Mar 18. Exp Neurol. 2009. PMID: 19303005 Review.
Cited by
-
Defective PTEN-induced kinase 1/Parkin mediated mitophagy and neurodegenerative diseases.Front Cell Neurosci. 2022 Oct 20;16:1031153. doi: 10.3389/fncel.2022.1031153. eCollection 2022. Front Cell Neurosci. 2022. PMID: 36339819 Free PMC article. Review.
-
High-content phenotyping of Parkinson's disease patient stem cell-derived midbrain dopaminergic neurons using machine learning classification.Stem Cell Reports. 2022 Oct 11;17(10):2349-2364. doi: 10.1016/j.stemcr.2022.09.001. Epub 2022 Sep 29. Stem Cell Reports. 2022. PMID: 36179692 Free PMC article.
-
Novel protein complexes containing autophagy and UPS components regulate proteasome-dependent PARK2 recruitment onto mitochondria and PARK2-PARK6 activity during mitophagy.Cell Death Dis. 2022 Nov 10;13(11):947. doi: 10.1038/s41419-022-05339-x. Cell Death Dis. 2022. PMID: 36357363 Free PMC article.
-
Mitophagy and reactive oxygen species interplay in Parkinson's disease.NPJ Parkinsons Dis. 2022 Oct 18;8(1):135. doi: 10.1038/s41531-022-00402-y. NPJ Parkinsons Dis. 2022. PMID: 36257956 Free PMC article. Review.
-
Mitophagy and Neurodegeneration: Between the Knowns and the Unknowns.Front Cell Dev Biol. 2022 Mar 22;10:837337. doi: 10.3389/fcell.2022.837337. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35392168 Free PMC article. Review.
References
-
- Berg D, Schweitzer KJ, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T, et al. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease*. Brain: a journal of neurology. 2005;128:3000–3011. - PubMed
-
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. - PubMed
-
- Bonifati V. Deciphering Parkinson’s disease--PARK8. The Lancet Neurology. 2002;1:83. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
