

VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington's disease
- PMID: 27561680
- PMCID: PMC5007466
- DOI: 10.1038/ncomms12646
VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington's disease
Abstract
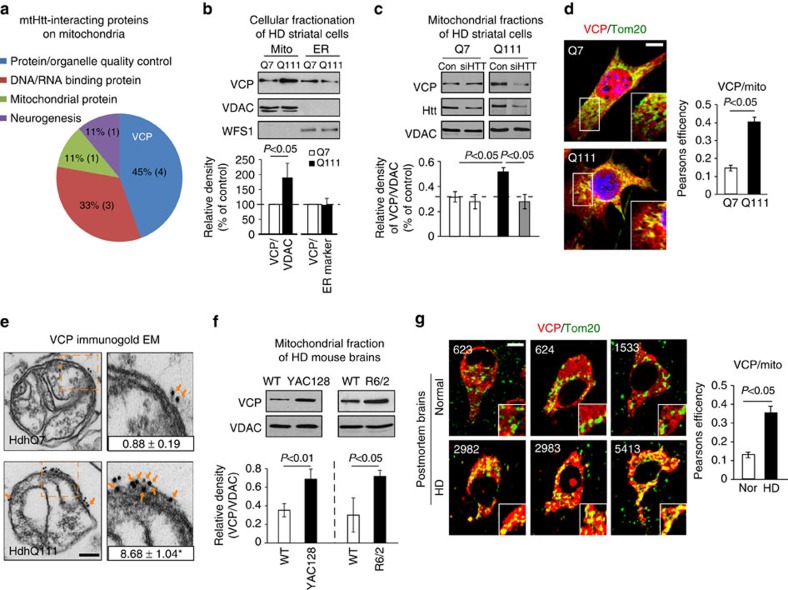
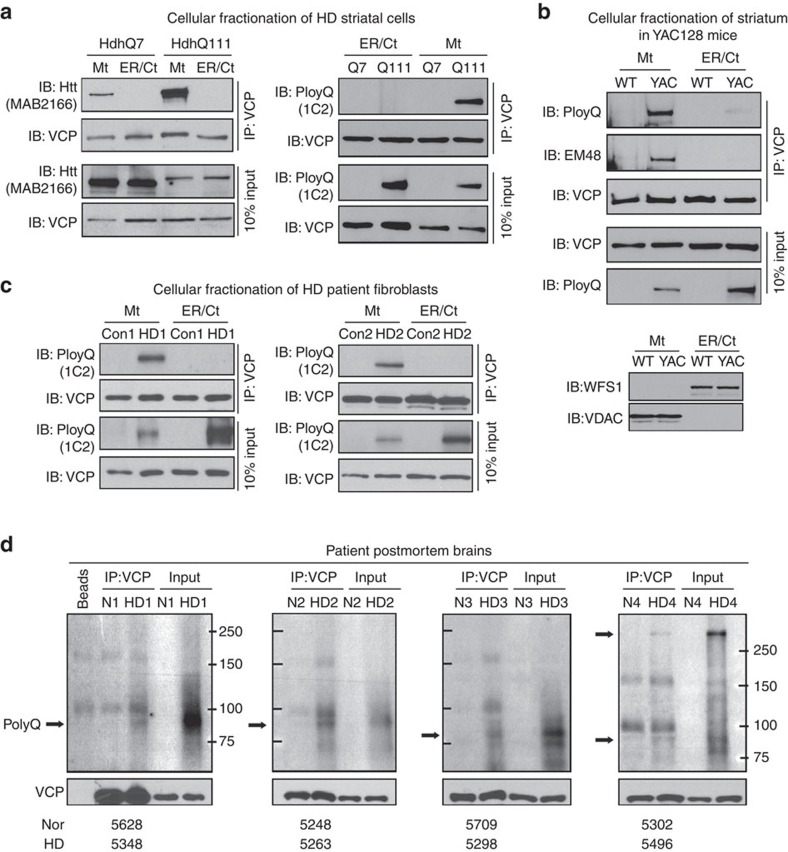
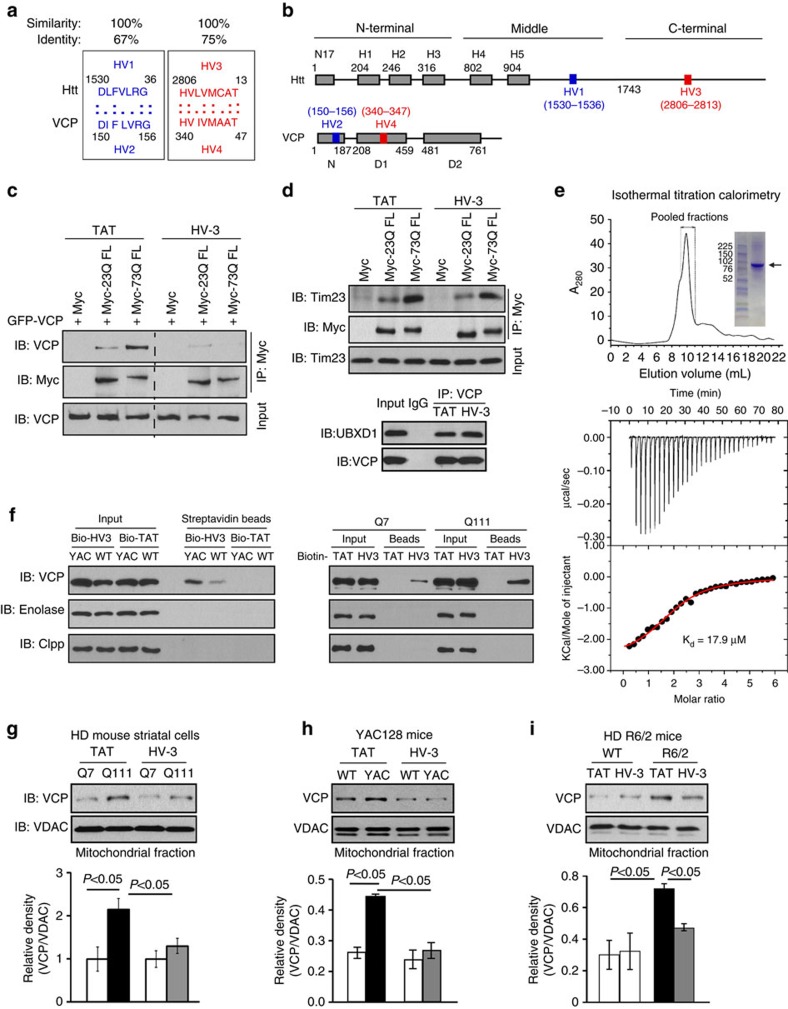
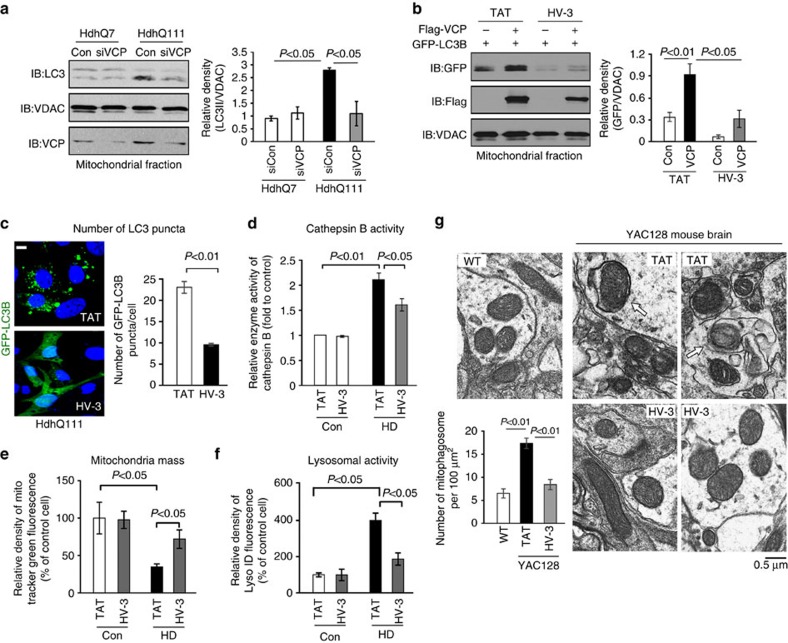
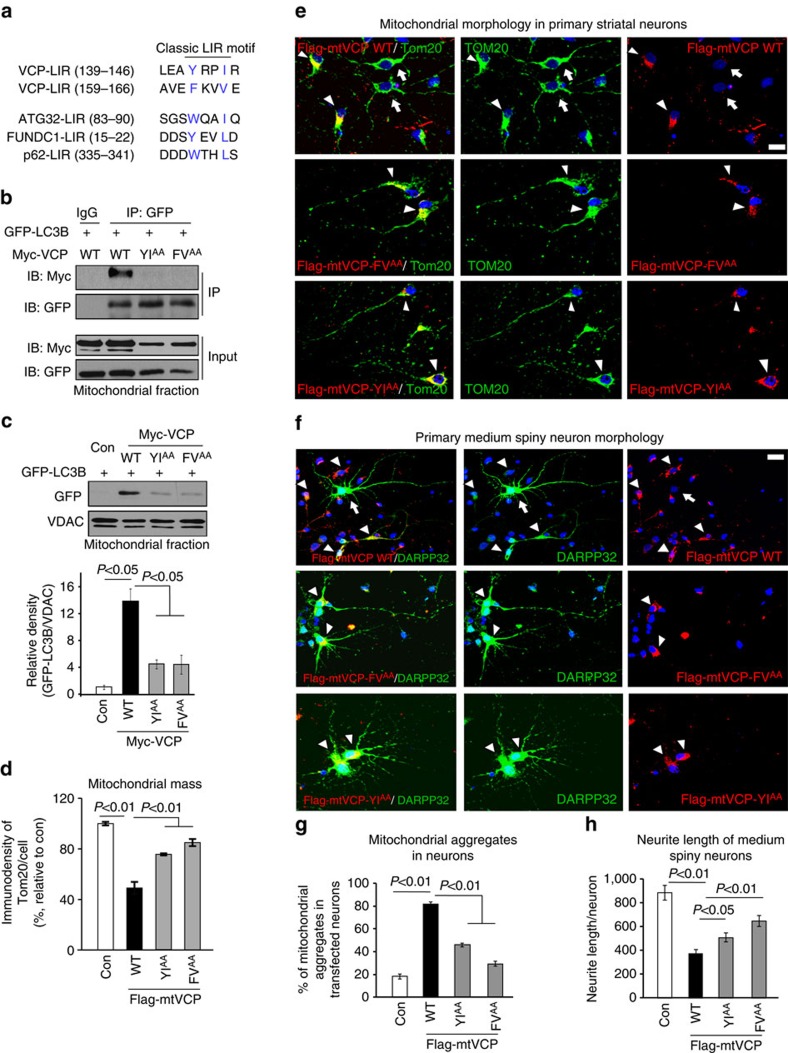
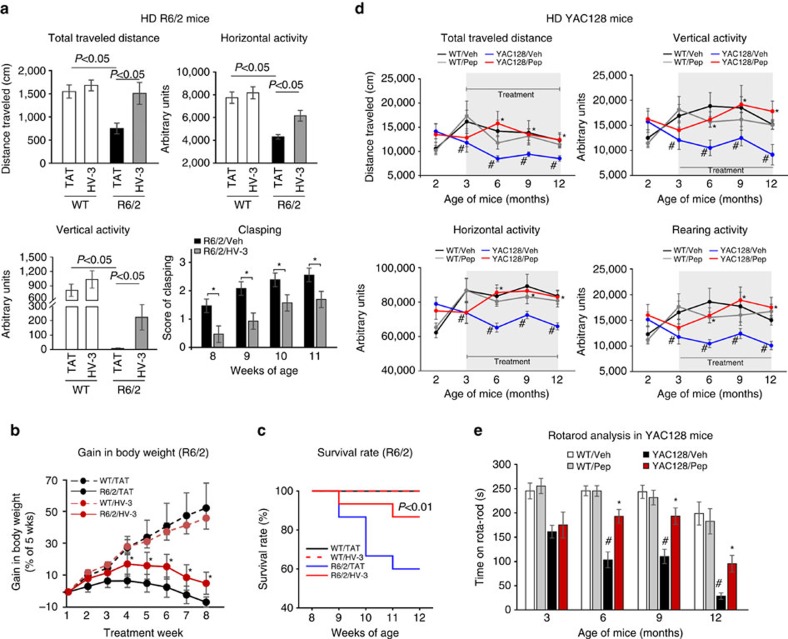
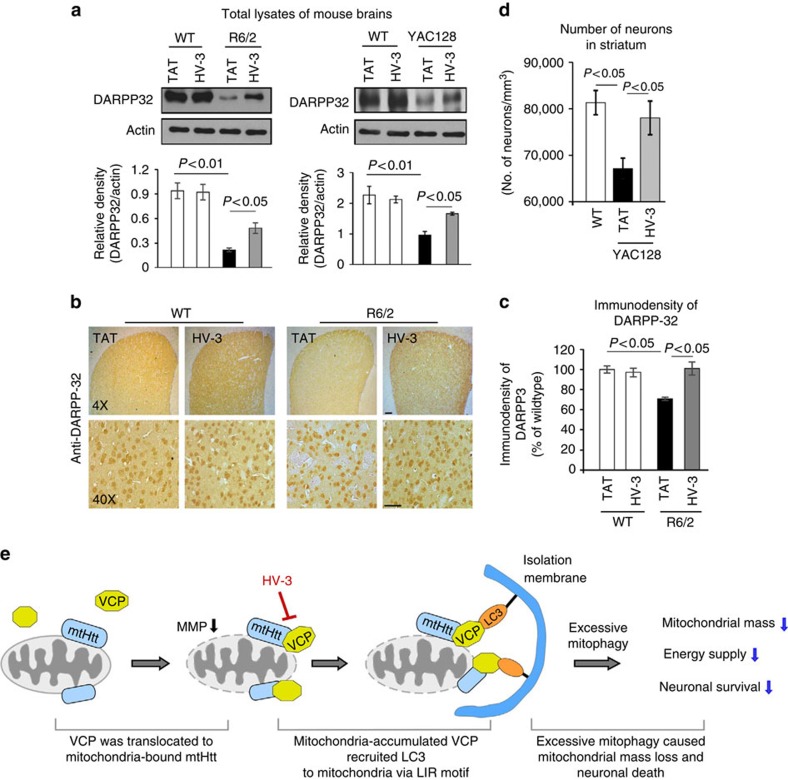
Mutant Huntingtin (mtHtt) causes neurodegeneration in Huntington's disease (HD) by evoking defects in the mitochondria, but the underlying mechanisms remains elusive. Our proteomic analysis identifies valosin-containing protein (VCP) as an mtHtt-binding protein on the mitochondria. Here we show that VCP is selectively translocated to the mitochondria, where it is bound to mtHtt in various HD models. Mitochondria-accumulated VCP elicits excessive mitophagy, causing neuronal cell death. Blocking mtHtt/VCP mitochondrial interaction with a peptide, HV-3, abolishes VCP translocation to the mitochondria, corrects excessive mitophagy and reduces cell death in HD mouse- and patient-derived cells and HD transgenic mouse brains. Treatment with HV-3 reduces behavioural and neuropathological phenotypes of HD in both fragment- and full-length mtHtt transgenic mice. Our findings demonstrate a causal role of mtHtt-induced VCP mitochondrial accumulation in HD pathogenesis and suggest that the peptide HV-3 might be a useful tool for developing new therapeutics to treat HD.
Conflict of interest statement
A patent on the design and applications of the HV-3 peptide inhibitor has been filed. The authors declare no conflict of interest.
Figures

Similar articles
-
VCP cooperates with UBXD1 to degrade mitochondrial outer membrane protein MCL1 in model of Huntington's disease.Biochim Biophys Acta Mol Basis Dis. 2017 Feb;1863(2):552-559. doi: 10.1016/j.bbadis.2016.11.026. Epub 2016 Nov 29. Biochim Biophys Acta Mol Basis Dis. 2017. PMID: 27913212 Free PMC article.
-
Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability.Biochim Biophys Acta Mol Basis Dis. 2019 Jun 1;1865(6):1428-1435. doi: 10.1016/j.bbadis.2019.02.015. Epub 2019 Feb 23. Biochim Biophys Acta Mol Basis Dis. 2019. PMID: 30802639 Free PMC article.
-
Inhibition of mitochondrial fragmentation diminishes Huntington's disease-associated neurodegeneration.J Clin Invest. 2013 Dec;123(12):5371-88. doi: 10.1172/JCI70911. Epub 2013 Nov 15. J Clin Invest. 2013. PMID: 24231356 Free PMC article.
-
Mitophagy in Huntington's disease.Neurochem Int. 2021 Oct;149:105147. doi: 10.1016/j.neuint.2021.105147. Epub 2021 Jul 28. Neurochem Int. 2021. PMID: 34329735 Review.
-
Mitochondrial Abnormalities and Synaptic Damage in Huntington's Disease: a Focus on Defective Mitophagy and Mitochondria-Targeted Therapeutics.Mol Neurobiol. 2021 Dec;58(12):6350-6377. doi: 10.1007/s12035-021-02556-x. Epub 2021 Sep 14. Mol Neurobiol. 2021. PMID: 34519969 Review.
Cited by
-
Localized K63 ubiquitin signaling is regulated by VCP/p97 during oxidative stress.bioRxiv [Preprint]. 2024 Jun 21:2024.06.20.598218. doi: 10.1101/2024.06.20.598218. bioRxiv. 2024. PMID: 38948861 Free PMC article. Preprint.
-
Phosphorylation of mitochondrial matrix proteins regulates their selective mitophagic degradation.Proc Natl Acad Sci U S A. 2019 Oct 8;116(41):20517-20527. doi: 10.1073/pnas.1901759116. Epub 2019 Sep 23. Proc Natl Acad Sci U S A. 2019. PMID: 31548421 Free PMC article.
-
Mechanisms and roles of mitophagy in neurodegenerative diseases.CNS Neurosci Ther. 2019 Jul;25(7):859-875. doi: 10.1111/cns.13140. Epub 2019 May 2. CNS Neurosci Ther. 2019. PMID: 31050206 Free PMC article. Review.
-
Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression.Autophagy. 2018;14(3):534-551. doi: 10.1080/15548627.2017.1385674. Epub 2017 Dec 17. Autophagy. 2018. PMID: 28980850 Free PMC article.
-
Mitochondrial membranes modify mutant huntingtin aggregation.Biochim Biophys Acta Biomembr. 2021 Oct 1;1863(10):183663. doi: 10.1016/j.bbamem.2021.183663. Epub 2021 Jun 2. Biochim Biophys Acta Biomembr. 2021. PMID: 34089719 Free PMC article.
References
-
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 (1993). - PubMed
-
- Graveland G. A., Williams R. S. & DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science 227, 770–773 (1985). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
