

Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability
- PMID: 27384655
- PMCID: PMC5008087
- DOI: 10.1128/JVI.00923-16
Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability
Abstract
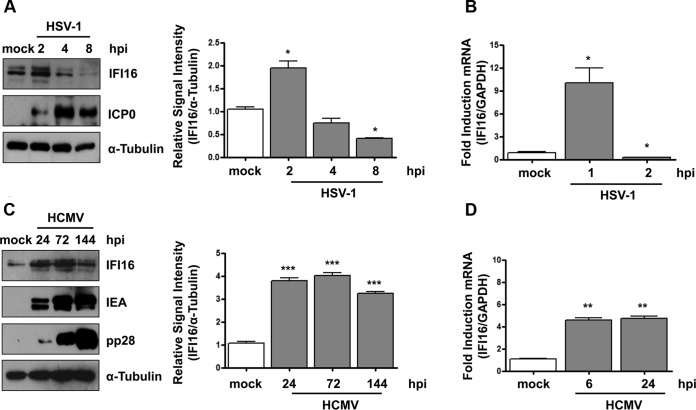
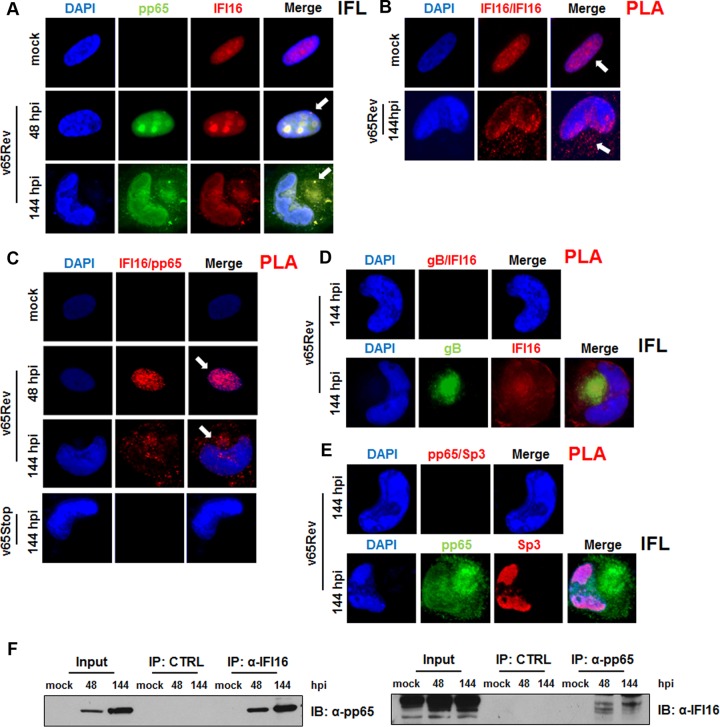
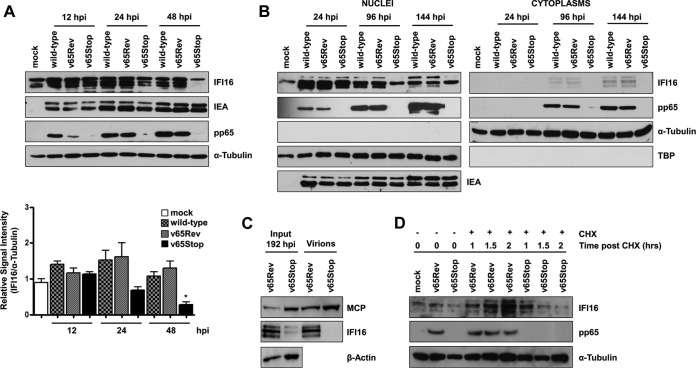
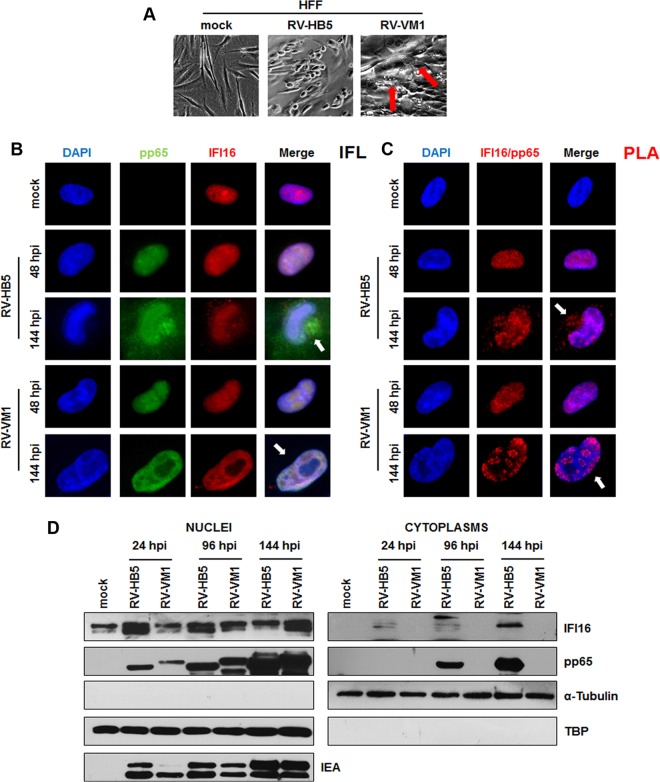
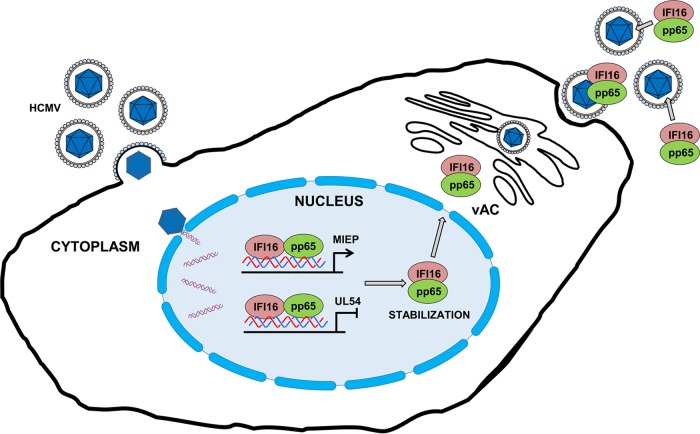
A key player in the intrinsic resistance against human cytomegalovirus (HCMV) is the interferon-γ-inducible protein 16 (IFI16), which behaves as a viral DNA sensor in the first hours postinfection and as a repressor of viral gene transcription in the later stages. Previous studies on HCMV replication demonstrated that IFI16 binds to the viral protein kinase pUL97, undergoes phosphorylation, and relocalizes to the cytoplasm of infected cells. In this study, we demonstrate that the tegument protein pp65 (pUL83) recruits IFI16 to the promoter of the UL54 gene and downregulates viral replication, as shown by use of the HCMV mutant v65Stop, which lacks pp65 expression. Interestingly, at late time points of HCMV infection, IFI16 is stabilized by its interaction with pp65, which stood in contrast to IFI16 degradation, observed in herpes simplex virus 1 (HSV-1)-infected cells. Moreover, we found that its translocation to the cytoplasm, in addition to pUL97, strictly depends on pp65, as demonstrated with the HCMV mutant RV-VM1, which expresses a form of pp65 unable to translocate into the cytoplasm. Thus, these data reveal a dual role for pp65: during early infection, it modulates IFI16 activity at the promoter of immediate-early and early genes; subsequently, it delocalizes IFI16 from the nucleus into the cytoplasm, thereby stabilizing and protecting it from degradation. Overall, these data identify a novel activity of the pp65/IFI16 interactome involved in the regulation of UL54 gene expression and IFI16 stability during early and late phases of HCMV replication.
Importance: The DNA sensor IFI16, a member of the PYHIN proteins, restricts HCMV replication by impairing viral DNA synthesis. Using a mutant virus lacking the tegument protein pp65 (v65Stop), we demonstrate that pp65 recruits IFI16 to the early UL54 gene promoter. As a putative counteraction to its restriction activity, pp65 supports the nucleocytoplasmic export of IFI16, which was demonstrated with the viral mutant RV-VM1 expressing a nuclearly retained pp65. These data reveal a dual role of pp65 in IFI16 regulation: in the early phase of HCMV infection, it contributes to viral evasion from IFI16 restriction activity, while at later time points, it promotes the nuclear delocalization of IFI16, thereby stabilizing and protecting it from degradation. In the present work, we further clarify the mechanisms HCMV relies on to overcome intracellular innate immune restriction and provide new insights into the relevance of DNA-sensing restriction factor IFI16 during HCMV infection.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage.J Virol. 2014 Jun;88(12):6970-82. doi: 10.1128/JVI.00384-14. Epub 2014 Apr 2. J Virol. 2014. PMID: 24696486 Free PMC article.
-
Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein.J Virol. 2010 Aug;84(15):7803-14. doi: 10.1128/JVI.00139-10. Epub 2010 May 26. J Virol. 2010. PMID: 20504932 Free PMC article.
-
The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication.PLoS Pathog. 2012 Jan;8(1):e1002498. doi: 10.1371/journal.ppat.1002498. Epub 2012 Jan 26. PLoS Pathog. 2012. PMID: 22291595 Free PMC article.
-
The human cytomegalovirus tegument protein pp65 (pUL83): a key player in innate immune evasion.New Microbiol. 2018 Apr;41(2):87-94. Epub 2018 Jan 31. New Microbiol. 2018. PMID: 29384558 Review.
-
Intrinsic host restriction factors of human cytomegalovirus replication and mechanisms of viral escape.World J Virol. 2016 Aug 12;5(3):87-96. doi: 10.5501/wjv.v5.i3.87. World J Virol. 2016. PMID: 27563536 Free PMC article. Review.
Cited by
-
Understanding HCMV Latency Using Unbiased Proteomic Analyses.Pathogens. 2020 Jul 20;9(7):590. doi: 10.3390/pathogens9070590. Pathogens. 2020. PMID: 32698381 Free PMC article. Review.
-
Human cytomegalovirus overcomes SAMHD1 restriction in macrophages via pUL97.Nat Microbiol. 2019 Dec;4(12):2260-2272. doi: 10.1038/s41564-019-0557-8. Epub 2019 Sep 23. Nat Microbiol. 2019. PMID: 31548682
-
Insights into the Transcriptome of Human Cytomegalovirus: A Comprehensive Review.Viruses. 2023 Aug 8;15(8):1703. doi: 10.3390/v15081703. Viruses. 2023. PMID: 37632045 Free PMC article. Review.
-
Challenging the Conventional Interpretation of HCMV Seronegativity.Microorganisms. 2021 Nov 18;9(11):2382. doi: 10.3390/microorganisms9112382. Microorganisms. 2021. PMID: 34835508 Free PMC article.
-
γδ T Cell-Mediated Immunity to Cytomegalovirus Infection.Front Immunol. 2017 Feb 9;8:105. doi: 10.3389/fimmu.2017.00105. eCollection 2017. Front Immunol. 2017. PMID: 28232834 Free PMC article. Review.
References
-
- Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2702–2772. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA, USA.
-
- Johnson WE. 2013. Rapid adversarial co-evolution of viruses and cellular restriction factors. Curr Top Microbiol Immunol 371:123–151. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
