

Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes
- PMID: 27230643
- PMCID: PMC4888901
- DOI: 10.1161/CIRCRESAHA.116.306923
Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes
Abstract
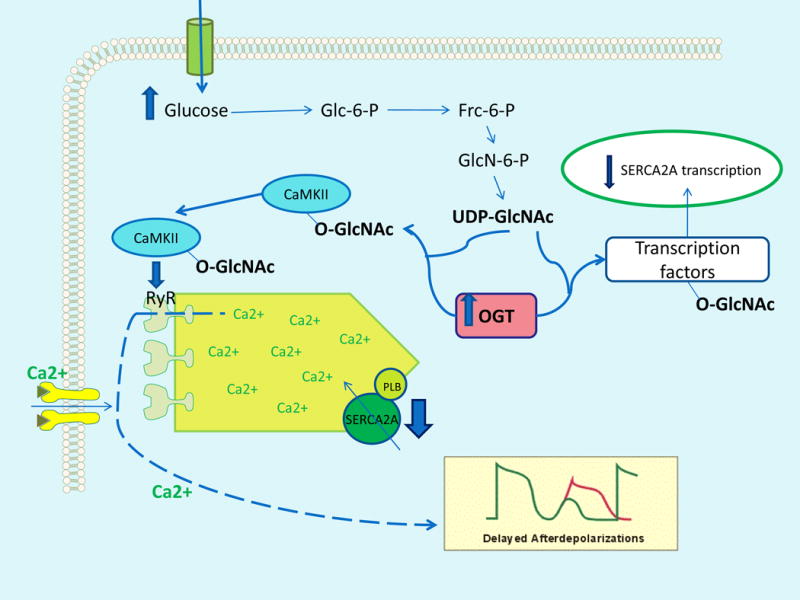
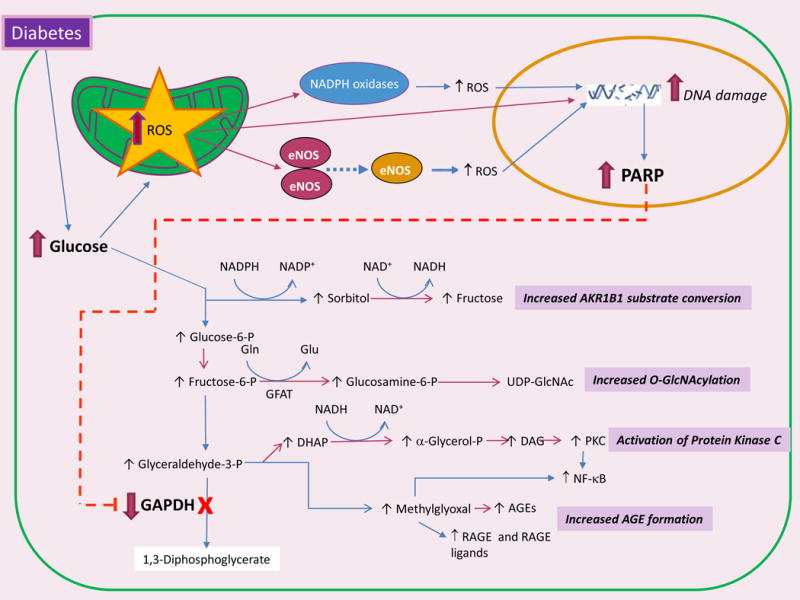
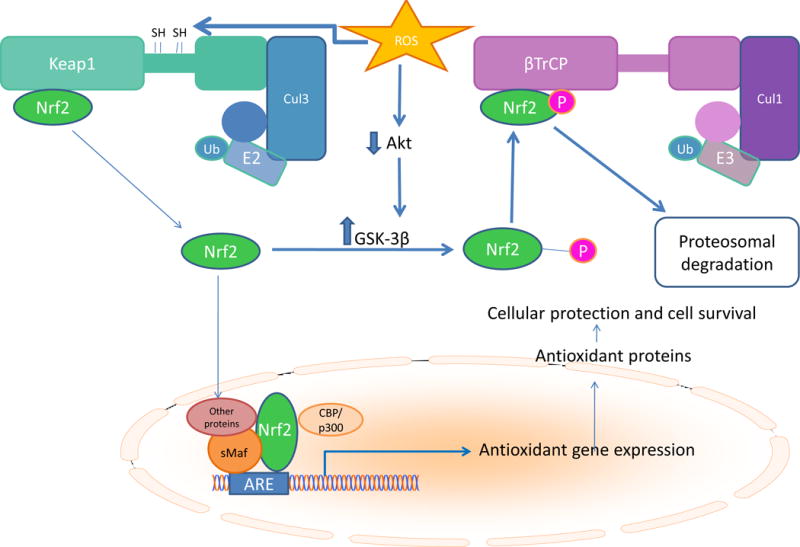
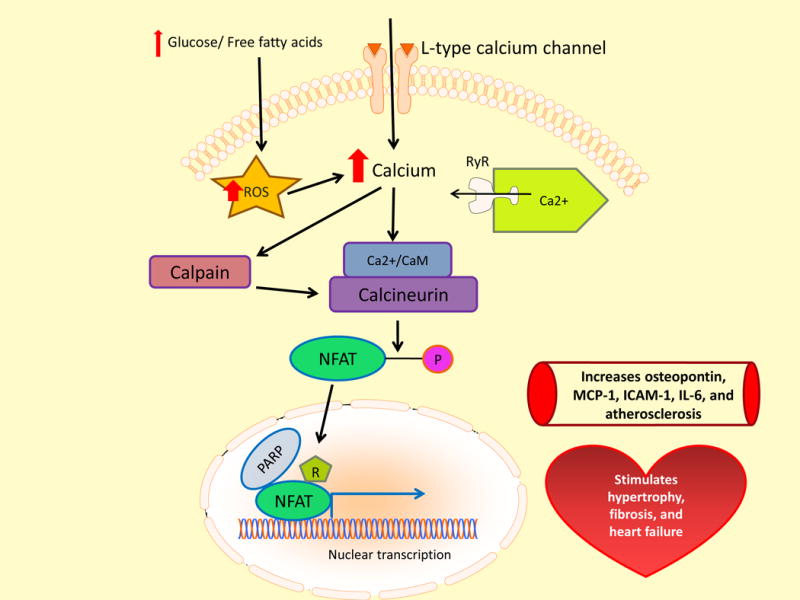
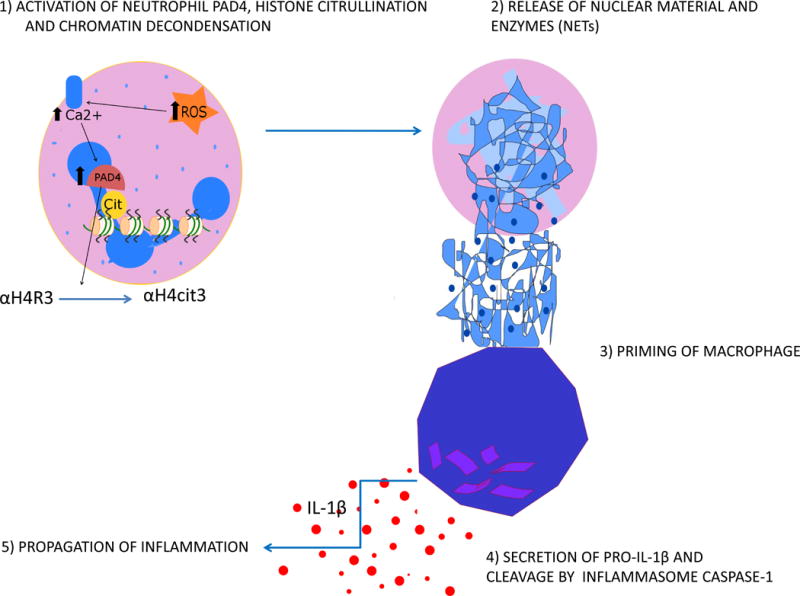
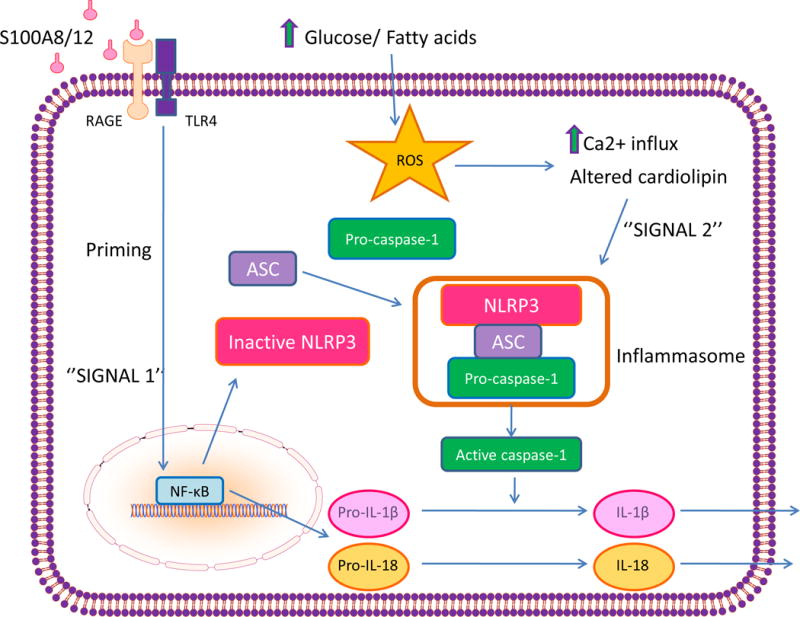
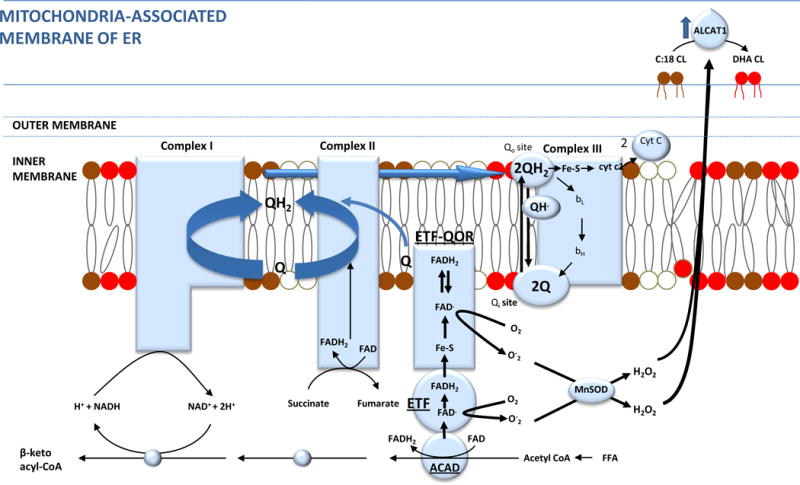
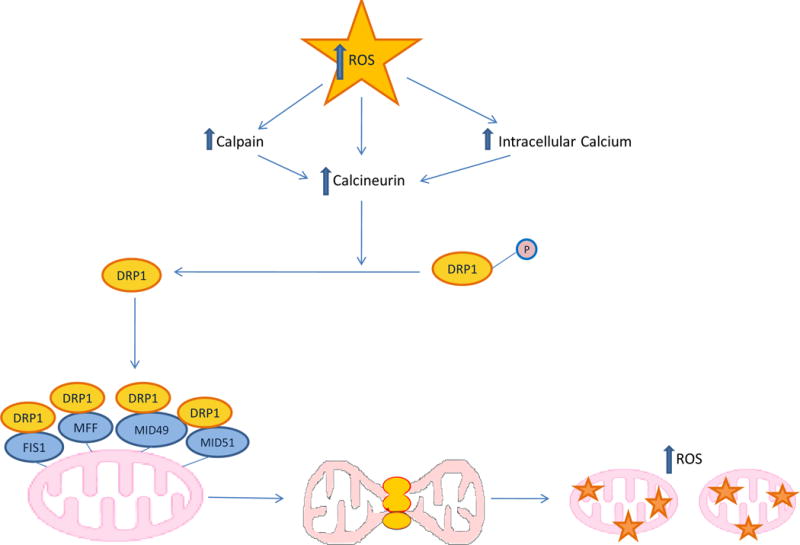
The clinical correlations linking diabetes mellitus with accelerated atherosclerosis, cardiomyopathy, and increased post-myocardial infarction fatality rates are increasingly understood in mechanistic terms. The multiple mechanisms discussed in this review seem to share a common element: prolonged increases in reactive oxygen species (ROS) production in diabetic cardiovascular cells. Intracellular hyperglycemia causes excessive ROS production. This activates nuclear poly(ADP-ribose) polymerase, which inhibits GAPDH, shunting early glycolytic intermediates into pathogenic signaling pathways. ROS and poly(ADP-ribose) polymerase also reduce sirtuin, PGC-1α, and AMP-activated protein kinase activity. These changes cause decreased mitochondrial biogenesis, increased ROS production, and disturbed circadian clock synchronization of glucose and lipid metabolism. Excessive ROS production also facilitates nuclear transport of proatherogenic transcription factors, increases transcription of the neutrophil enzyme initiating NETosis, peptidylarginine deiminase 4, and activates the NOD-like receptor family, pyrin domain-containing 3 inflammasome. Insulin resistance causes excessive cardiomyocyte ROS production by increasing fatty acid flux and oxidation. This stimulates overexpression of the nuclear receptor PPARα and nuclear translocation of forkhead box O 1, which cause cardiomyopathy. ROS also shift the balance between mitochondrial fusion and fission in favor of increased fission, reducing the metabolic capacity and efficiency of the mitochondrial electron transport chain and ATP synthesis. Mitochondrial oxidative stress also plays a central role in angiotensin II-induced gap junction remodeling and arrhythmogenesis. ROS contribute to sudden death in diabetics after myocardial infarction by increasing post-translational protein modifications, which cause increased ryanodine receptor phosphorylation and downregulation of sarco-endoplasmic reticulum Ca(++)-ATPase transcription. Increased ROS also depress autonomic ganglion synaptic transmission by oxidizing the nAch receptor α3 subunit, potentially contributing to the increased risk of fatal cardiac arrhythmias associated with diabetic cardiac autonomic neuropathy.
Keywords: atherosclerosis; diabetes mellitus; heart failure; insulin resistance; myocardial infarction; reactive oxygen species.
© 2016 American Heart Association, Inc.
Figures
Similar articles
-
An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy.Int J Cardiol. 2013 Oct 9;168(4):3160-72. doi: 10.1016/j.ijcard.2013.07.150. Epub 2013 Aug 6. Int J Cardiol. 2013. PMID: 23932046 Review.
-
Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders.Cell Signal. 2013 Oct;25(10):1939-48. doi: 10.1016/j.cellsig.2013.06.007. Epub 2013 Jun 11. Cell Signal. 2013. PMID: 23770291 Review.
-
Myocardial infarction in rats causes partial impairment in insulin response associated with reduced fatty acid oxidation and mitochondrial gene expression.J Thorac Cardiovasc Surg. 2010 Nov;140(5):1160-7. doi: 10.1016/j.jtcvs.2010.08.003. Epub 2010 Sep 17. J Thorac Cardiovasc Surg. 2010. PMID: 20850803
-
Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling contributing to increased vulnerability in diabetic heart.Basic Res Cardiol. 2013 May;108(3):329. doi: 10.1007/s00395-013-0329-1. Epub 2013 Mar 5. Basic Res Cardiol. 2013. PMID: 23460046
-
Oxidative stress and diabetic complications.Circ Res. 2010 Oct 29;107(9):1058-70. doi: 10.1161/CIRCRESAHA.110.223545. Circ Res. 2010. PMID: 21030723 Free PMC article. Review.
Cited by
-
Elevated levels of urine isocitrate, hydroxymethylglutarate, and formiminoglutamate are associated with arterial stiffness in Korean adults.Sci Rep. 2021 May 13;11(1):10180. doi: 10.1038/s41598-021-89639-w. Sci Rep. 2021. PMID: 33986342 Free PMC article.
-
Endothelial Senescence: From Macro- to Micro-Vasculature and Its Implications on Cardiovascular Health.Int J Mol Sci. 2024 Feb 6;25(4):1978. doi: 10.3390/ijms25041978. Int J Mol Sci. 2024. PMID: 38396653 Free PMC article. Review.
-
The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders.Cells. 2020 Jul 30;9(8):1812. doi: 10.3390/cells9081812. Cells. 2020. PMID: 32751658 Free PMC article. Review.
-
Endothelial dysfunction: molecular mechanisms and clinical implications.MedComm (2020). 2024 Jul 22;5(8):e651. doi: 10.1002/mco2.651. eCollection 2024 Aug. MedComm (2020). 2024. PMID: 39040847 Free PMC article. Review.
-
Advanced Glycation End Products: Potential Mechanism and Therapeutic Target in Cardiovascular Complications under Diabetes.Oxid Med Cell Longev. 2019 Dec 6;2019:9570616. doi: 10.1155/2019/9570616. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31885827 Free PMC article. Review.
References
-
- Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care. 1998;21:1138–45. - PubMed
-
- Gu K, Cowie CC, Harris MI. Diabetes and decline in heart disease mortality in US adults. Jama. 1999;281:1291–7. - PubMed
-
- Donahoe SM, Stewart GC, McCabe CH, Mohanavelu S, Murphy SA, Cannon CP, Antman EM. Diabetes and mortality following acute coronary syndromes. Jama. 2007;298:765–75. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
