

Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK
- PMID: 27193833
- PMCID: PMC4873974
- DOI: 10.1038/ncomms11363
Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK
Abstract
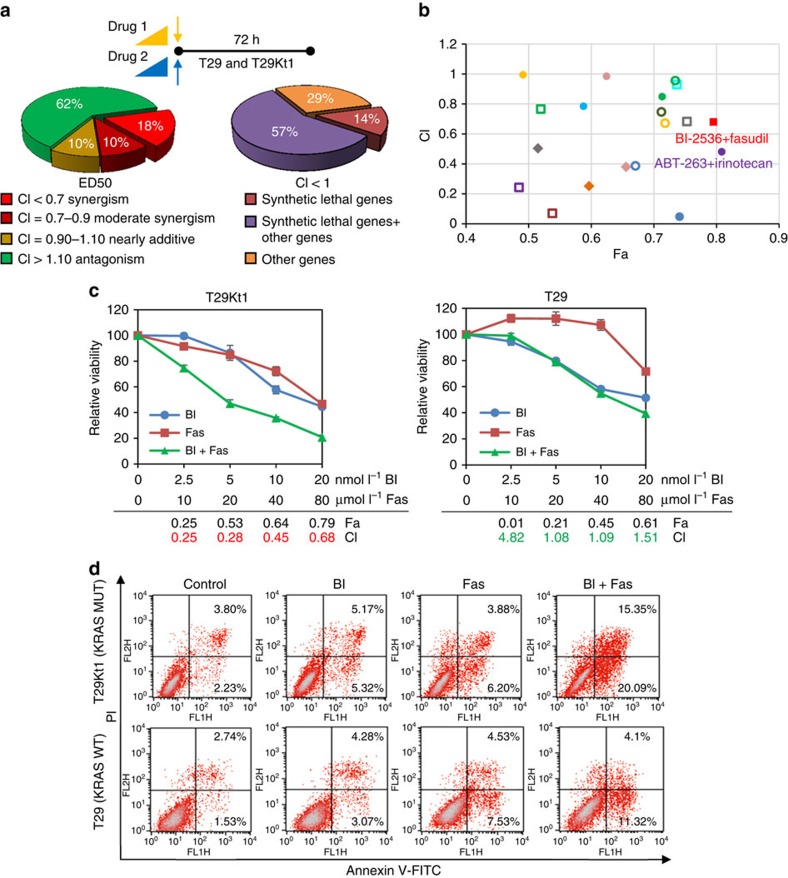
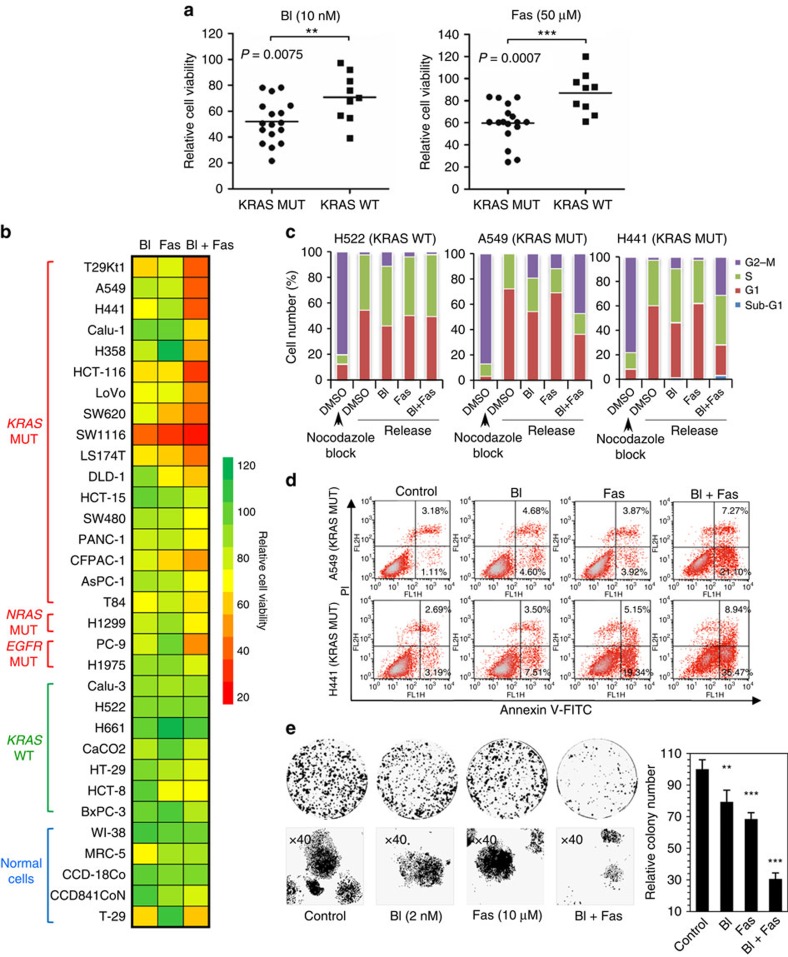
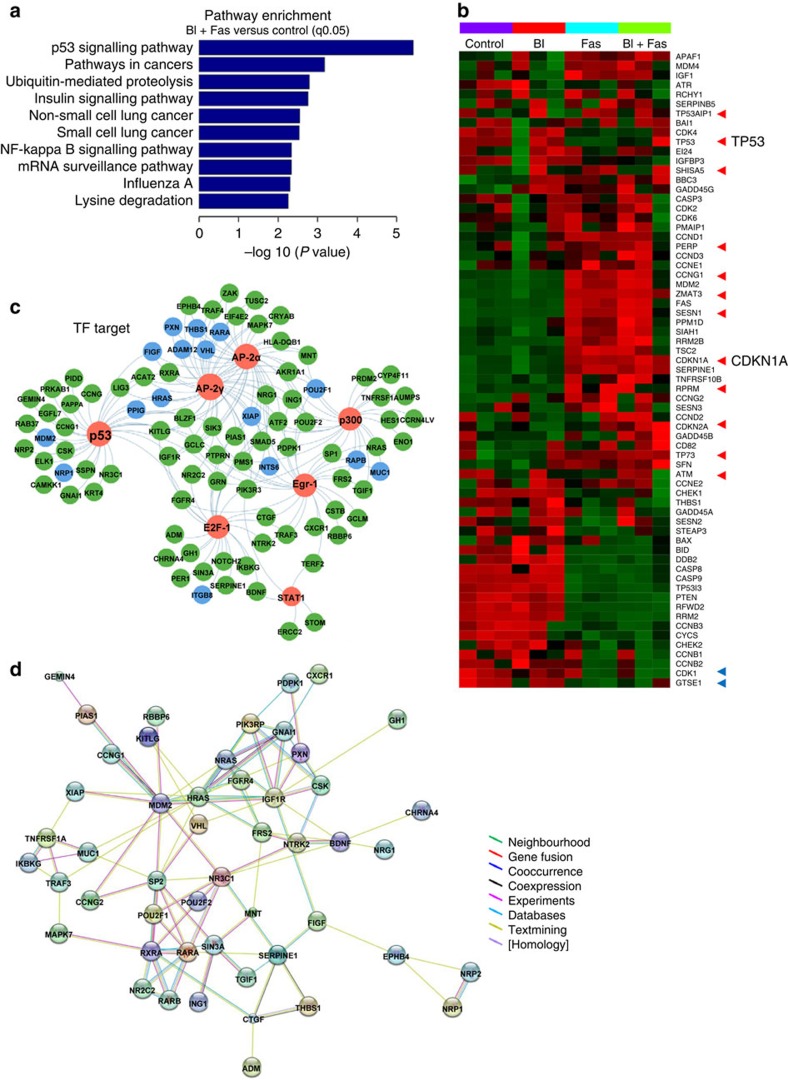
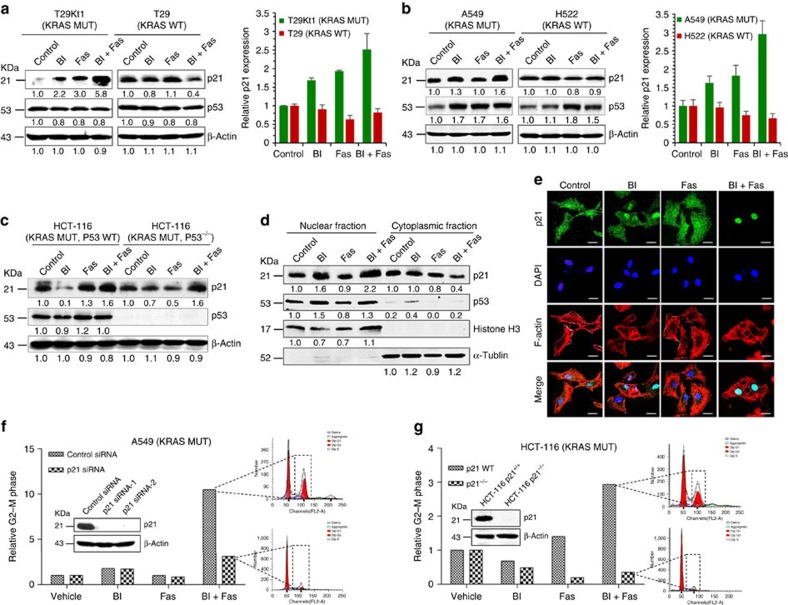
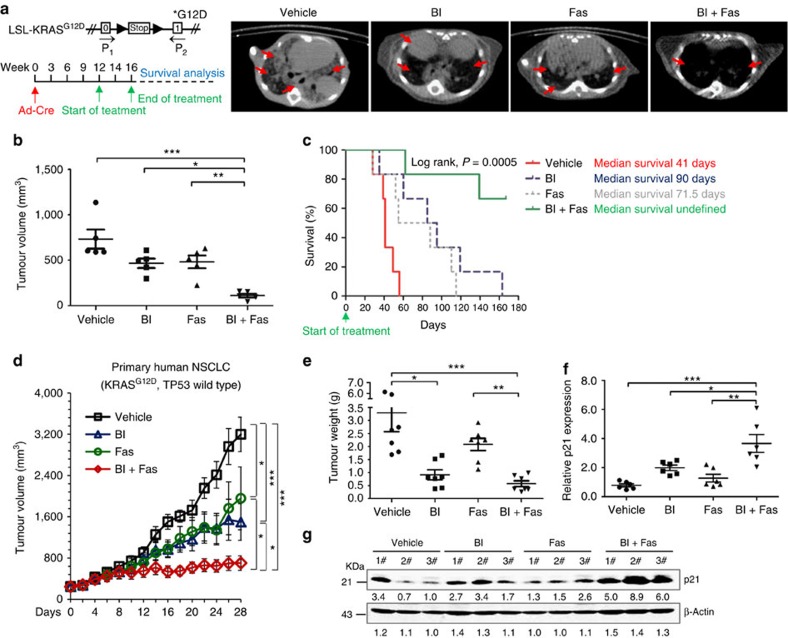
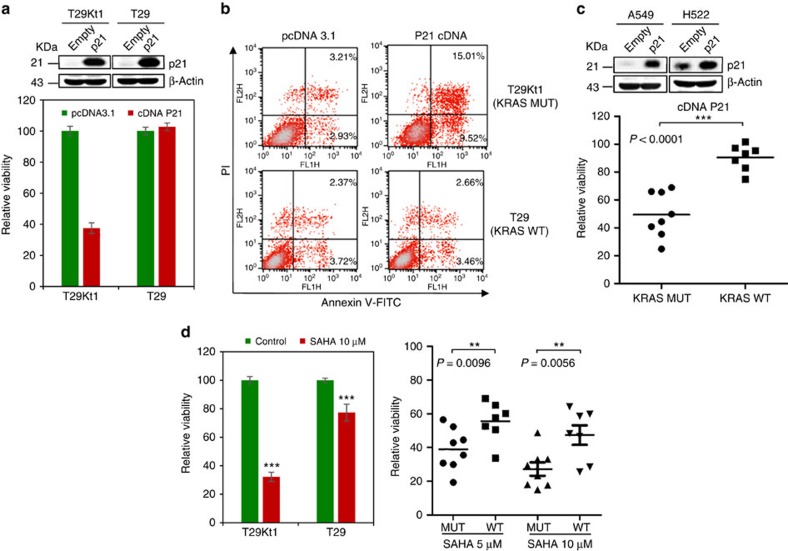
No effective targeted therapies exist for cancers with somatic KRAS mutations. Here we develop a synthetic lethal chemical screen in isogenic KRAS-mutant and wild-type cells to identify clinical drug pairs. Our results show that dual inhibition of polo-like kinase 1 and RhoA/Rho kinase (ROCK) leads to the synergistic effects in KRAS-mutant cancers. Microarray analysis reveals that this combinatory inhibition significantly increases transcription and activity of cyclin-dependent kinase inhibitor p21(WAF1/CIP1), leading to specific G2/M phase blockade in KRAS-mutant cells. Overexpression of p21(WAF1/CIP1), either by cDNA transfection or clinical drugs, preferentially impairs the growth of KRAS-mutant cells, suggesting a druggable synthetic lethal interaction between KRAS and p21(WAF1/CIP1). Co-administration of BI-2536 and fasudil either in the LSL-KRAS(G12D) mouse model or in a patient tumour explant mouse model of KRAS-mutant lung cancer suppresses tumour growth and significantly prolongs mouse survival, suggesting a strong synergy in vivo and a potential avenue for therapeutic treatment of KRAS-mutant cancers.
Figures
Similar articles
-
A combination therapy for KRAS-mutant lung cancer by targeting synthetic lethal partners of mutant KRAS.Chin J Cancer. 2016 Oct 28;35(1):92. doi: 10.1186/s40880-016-0154-7. Chin J Cancer. 2016. PMID: 27793187 Free PMC article.
-
Suppression of Polo like kinase 1 (PLK1) by p21(Waf1) mediates the p53-dependent prevention of caspase-independent mitotic death.Cell Signal. 2011 Nov;23(11):1816-23. doi: 10.1016/j.cellsig.2011.06.016. Epub 2011 Jun 29. Cell Signal. 2011. PMID: 21726628
-
Long-term downregulation of Polo-like kinase 1 increases the cyclin-dependent kinase inhibitor p21(WAF1/CIP1).Cell Cycle. 2009 Feb 1;8(3):460-72. doi: 10.4161/cc.8.3.7651. Epub 2009 Feb 18. Cell Cycle. 2009. PMID: 19177004
-
Molecular interactions of polo-like kinase 1 in human cancers.J Clin Pathol. 2016 Jul;69(7):557-62. doi: 10.1136/jclinpath-2016-203656. Epub 2016 Mar 3. J Clin Pathol. 2016. PMID: 26941182 Review.
-
Defeat mutant KRAS with synthetic lethality.Small GTPases. 2017 Oct 2;8(4):212-219. doi: 10.1080/21541248.2016.1213783. Epub 2016 Jul 27. Small GTPases. 2017. PMID: 27463838 Free PMC article. Review.
Cited by
-
KRAS and MET in non-small-cell lung cancer: two of the new kids on the 'drivers' block.Ther Adv Respir Dis. 2022 Jan-Dec;16:17534666211066064. doi: 10.1177/17534666211066064. Ther Adv Respir Dis. 2022. PMID: 35098800 Free PMC article. Review.
-
Rho kinase proteins display aberrant upregulation in vascular tumors and contribute to vascular tumor growth.BMC Cancer. 2017 Jul 14;17(1):485. doi: 10.1186/s12885-017-3470-7. BMC Cancer. 2017. PMID: 28709411 Free PMC article.
-
An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer.Nat Commun. 2017 Feb 21;8:14294. doi: 10.1038/ncomms14294. Nat Commun. 2017. PMID: 28220783 Free PMC article.
-
BRCA1 orchestrates the response to BI-2536 and its combination with alisertib in MYC-driven small cell lung cancer.Cell Death Dis. 2024 Jul 31;15(7):551. doi: 10.1038/s41419-024-06950-w. Cell Death Dis. 2024. PMID: 39085197 Free PMC article.
-
Pharmacological inhibition of dihydroorotate dehydrogenase induces apoptosis and differentiation in acute myeloid leukemia cells.Haematologica. 2018 Sep;103(9):1472-1483. doi: 10.3324/haematol.2018.188185. Epub 2018 Jun 7. Haematologica. 2018. PMID: 29880605 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
