

A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2- Metastatic Breast Cancer
- PMID: 27126994
- PMCID: PMC5085926
- DOI: 10.1158/1078-0432.CCR-16-0134
A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2- Metastatic Breast Cancer
Abstract
Purpose: Alpelisib, a selective oral inhibitor of the class I PI3K catalytic subunit p110α, has shown synergistic antitumor activity with endocrine therapy against ER+/PIK3CA-mutated breast cancer cells. This phase Ib study evaluated alpelisib plus letrozole's safety, tolerability, and preliminary activity in patients with metastatic ER+ breast cancer refractory to endocrine therapy.
Experimental design: Twenty-six patients received letrozole and alpelisib daily. Outcomes were assessed by standard solid-tumor phase I methods. Tumor blocks were collected for DNA extraction and next-generation sequencing.
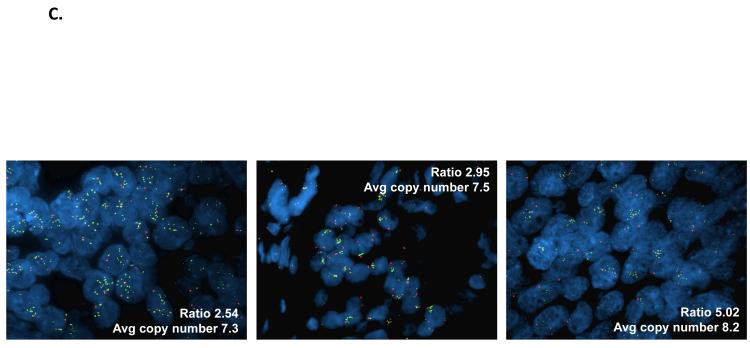
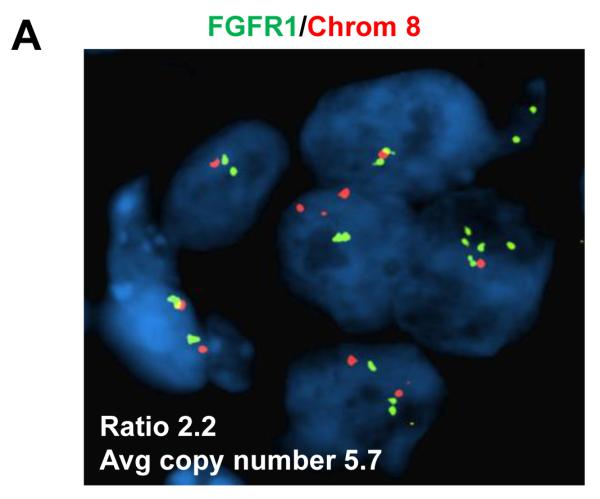
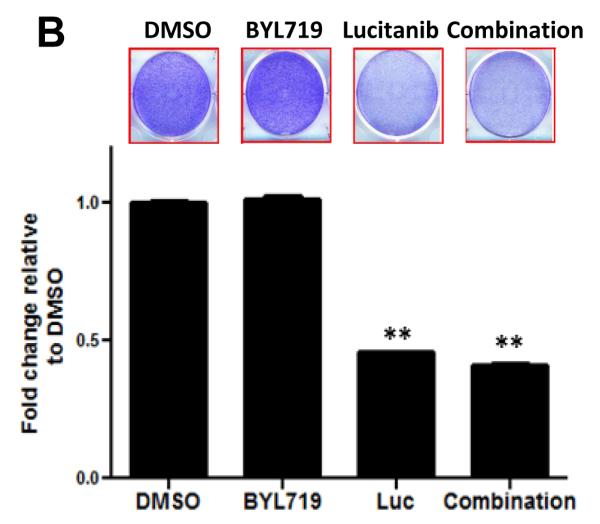
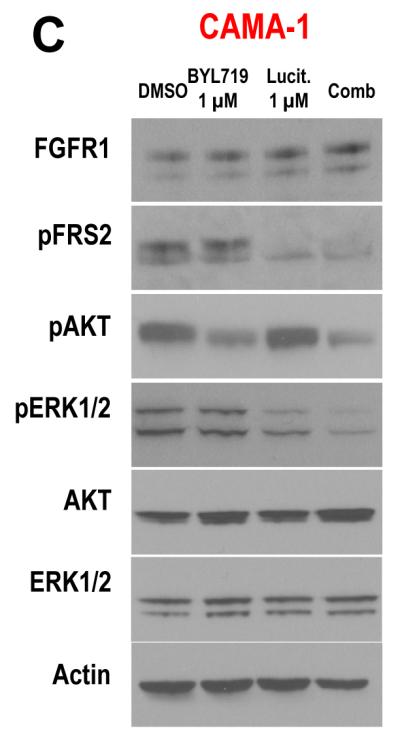
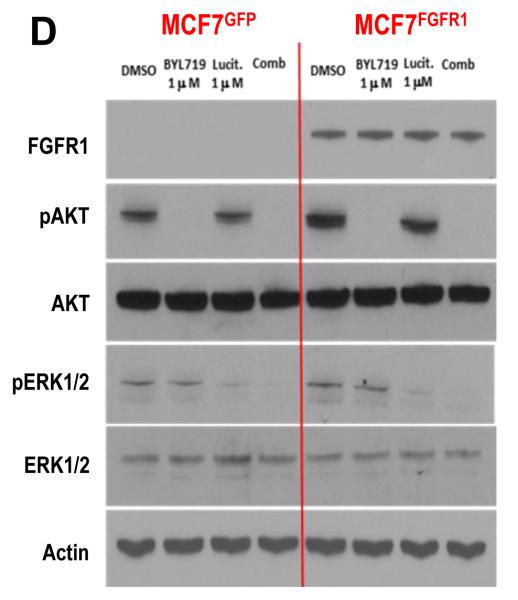
Results: Alpelisib's maximum-tolerated dose (MTD) in combination with letrozole was 300 mg/d. Common drug-related adverse events included hyperglycemia, nausea, fatigue, diarrhea, and rash with dose-limiting toxicity occurring at 350 mg/d of alpelisib. The clinical benefit rate (lack of progression ≥6 months) was 35% (44% in patients with PIK3CA-mutated and 20% in PIK3CA wild-type tumors; 95% CI, 17%-56%), including five objective responses. Of eight patients remaining on treatment ≥12 months, six had tumors with a PIK3CA mutation. Among evaluable tumors, those with FGFR1/2 amplification and KRAS and TP53 mutations did not derive clinical benefit. Overexpression of FGFR1 in ER+/PIK3CA mutant breast cancer cells attenuated the response to alpelisib in vitro CONCLUSIONS: The combination of letrozole and alpelisib was safe, with reversible toxicities. Clinical activity was observed independently of PIK3CA mutation status, although clinical benefit was seen in a higher proportion of patients with PIK3CA-mutated tumors. Phase II and III trials of alpelisib and endocrine therapy in patients with ER+ breast cancer are ongoing. Clin Cancer Res; 23(1); 26-34. ©2016 AACR.
©2016 American Association for Cancer Research.
Figures




Similar articles
-
Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer: A Phase 1b Clinical Trial.JAMA Oncol. 2019 Feb 1;5(2):e184475. doi: 10.1001/jamaoncol.2018.4475. Epub 2019 Feb 14. JAMA Oncol. 2019. PMID: 30543347 Free PMC article. Clinical Trial.
-
Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer.J Clin Oncol. 2014 Apr 20;32(12):1202-9. doi: 10.1200/JCO.2013.54.0518. Epub 2014 Mar 24. J Clin Oncol. 2014. PMID: 24663045 Free PMC article. Clinical Trial.
-
A Phase II Randomized Study of Neoadjuvant Letrozole Plus Alpelisib for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer (NEO-ORB).Clin Cancer Res. 2019 May 15;25(10):2975-2987. doi: 10.1158/1078-0432.CCR-18-3160. Epub 2019 Feb 5. Clin Cancer Res. 2019. PMID: 30723140 Free PMC article. Clinical Trial.
-
Everolimus versus alpelisib in advanced hormone receptor-positive HER2-negative breast cancer: targeting different nodes of the PI3K/AKT/mTORC1 pathway with different clinical implications.Breast Cancer Res. 2020 Apr 6;22(1):33. doi: 10.1186/s13058-020-01271-0. Breast Cancer Res. 2020. PMID: 32252811 Free PMC article. Review.
-
Alpelisib to treat breast cancer.Drugs Today (Barc). 2020 Jun;56(6):357-363. doi: 10.1358/dot.2020.56.6.3137526. Drugs Today (Barc). 2020. PMID: 32525134 Review.
Cited by
-
A PNPLA3-Deficient iPSC-Derived Hepatocyte Screen Identifies Pathways to Potentially Reduce Steatosis in Metabolic Dysfunction-Associated Fatty Liver Disease.Int J Mol Sci. 2024 Jul 2;25(13):7277. doi: 10.3390/ijms25137277. Int J Mol Sci. 2024. PMID: 39000384 Free PMC article.
-
Novel targeted therapies for metastatic breast cancer.Ann Transl Med. 2020 Jul;8(14):907. doi: 10.21037/atm.2020.03.43. Ann Transl Med. 2020. PMID: 32793751 Free PMC article. Review.
-
Management of toxicity to isoform α-specific PI3K inhibitors.Ann Oncol. 2019 Dec 1;30(Suppl_10):x21-x26. doi: 10.1093/annonc/mdz440. Ann Oncol. 2019. PMID: 31626273 Free PMC article. Review.
-
The overexpression of ZWINT in integrated bioinformatics analysis forecasts poor prognosis in breast cancer.Transl Cancer Res. 2020 Jan;9(1):187-193. doi: 10.21037/tcr.2019.12.66. Transl Cancer Res. 2020. PMID: 35117172 Free PMC article.
-
Successful Treatment of Hypoglycemia With Alpelisib in Pediatric Patients With PIK3CA-Related Overgrowth Spectrum.JCEM Case Rep. 2023 Apr 21;1(2):luad027. doi: 10.1210/jcemcr/luad027. eCollection 2023 Mar. JCEM Case Rep. 2023. PMID: 37908459 Free PMC article.
References
-
- Samuels Y, Diaz LA, Jr., Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7(6):561–73. - PubMed
-
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65(23):10992–1000. - PubMed
-
- Perez-Tenorio G, Alkhori L, Olsson B, Waltersson MA, Nordenskjold B, Rutqvist LE, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007;13(12):3577–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
