

Simulated binding of transcription factors to active and inactive regions folds human chromosomes into loops, rosettes and topological domains
- PMID: 27060145
- PMCID: PMC4856988
- DOI: 10.1093/nar/gkw135
Simulated binding of transcription factors to active and inactive regions folds human chromosomes into loops, rosettes and topological domains
Abstract
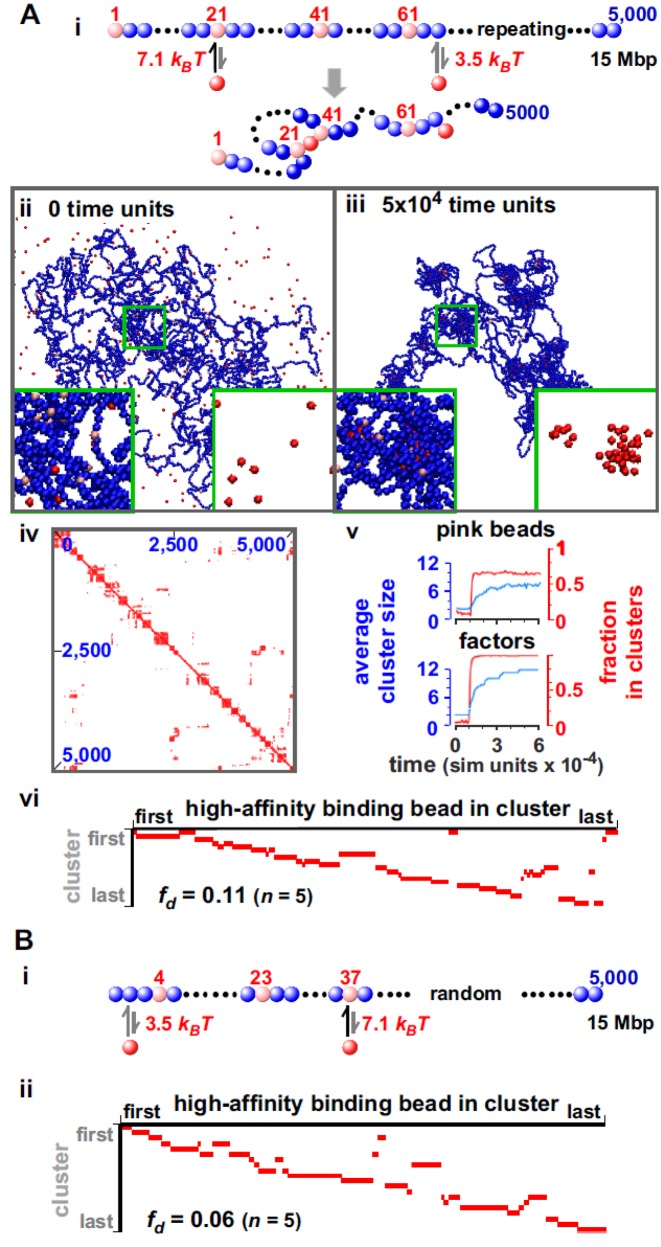
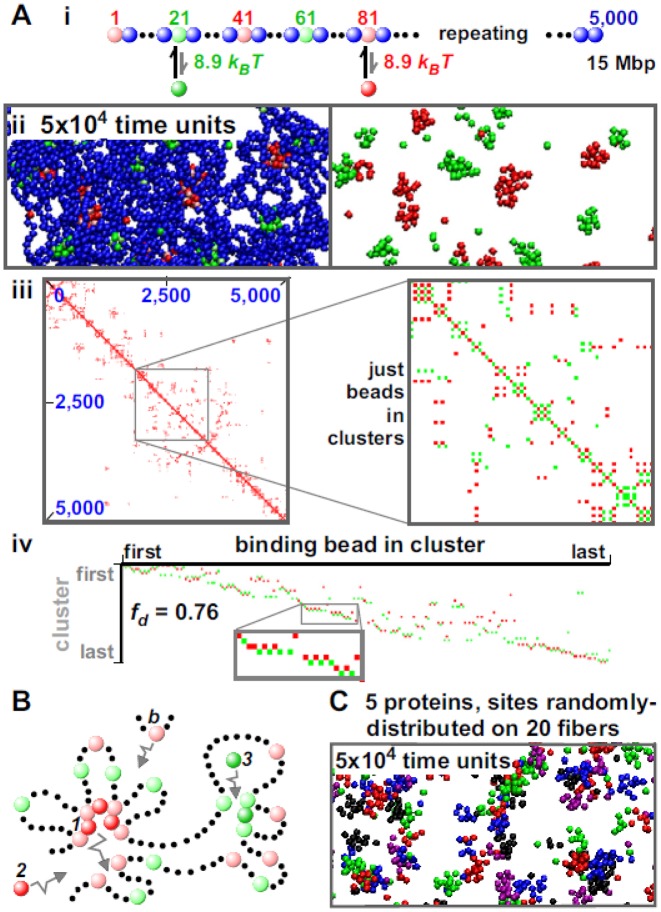
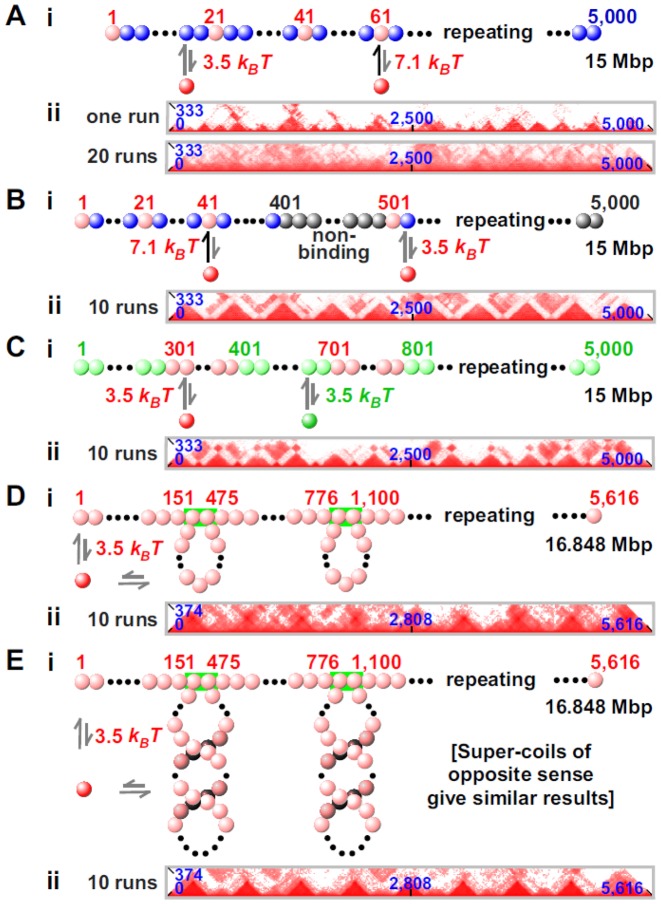
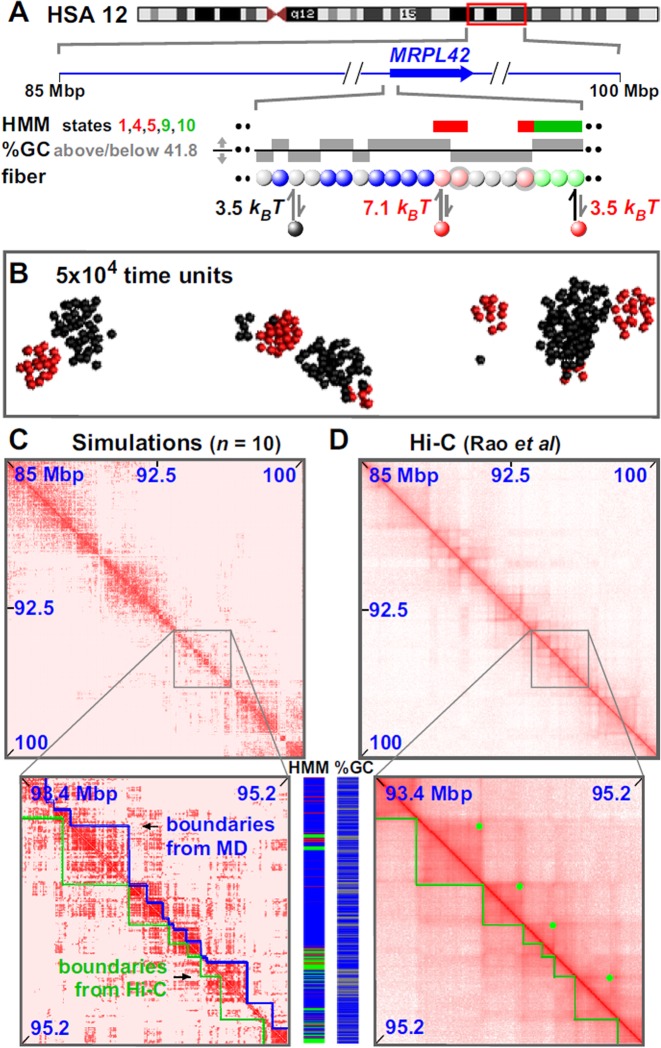
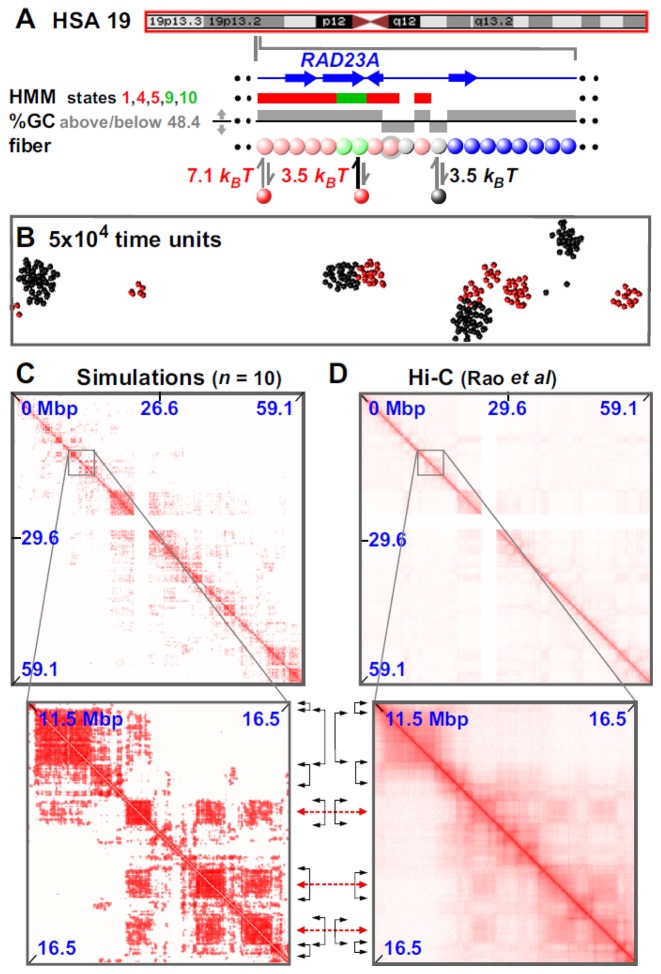
Biophysicists are modeling conformations of interphase chromosomes, often basing the strengths of interactions between segments distant on the genetic map on contact frequencies determined experimentally. Here, instead, we develop a fitting-free, minimal model: bivalent or multivalent red and green 'transcription factors' bind to cognate sites in strings of beads ('chromatin') to form molecular bridges stabilizing loops. In the absence of additional explicit forces, molecular dynamic simulations reveal that bound factors spontaneously cluster-red with red, green with green, but rarely red with green-to give structures reminiscent of transcription factories. Binding of just two transcription factors (or proteins) to active and inactive regions of human chromosomes yields rosettes, topological domains and contact maps much like those seen experimentally. This emergent 'bridging-induced attraction' proves to be a robust, simple and generic force able to organize interphase chromosomes at all scales.
© The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Simulating topological domains in human chromosomes with a fitting-free model.Nucleus. 2016 Sep 2;7(5):453-461. doi: 10.1080/19491034.2016.1239684. Nucleus. 2016. PMID: 27841970 Free PMC article.
-
Simulation of different three-dimensional polymer models of interphase chromosomes compared to experiments-an evaluation and review framework of the 3D genome organization.Semin Cell Dev Biol. 2019 Jun;90:19-42. doi: 10.1016/j.semcdb.2018.07.012. Epub 2018 Aug 24. Semin Cell Dev Biol. 2019. PMID: 30125668 Review.
-
Nonspecific bridging-induced attraction drives clustering of DNA-binding proteins and genome organization.Proc Natl Acad Sci U S A. 2013 Sep 17;110(38):E3605-11. doi: 10.1073/pnas.1302950110. Epub 2013 Sep 3. Proc Natl Acad Sci U S A. 2013. PMID: 24003126 Free PMC article.
-
Multi-contact 3C reveals that the human genome during interphase is largely not entangled.Nat Struct Mol Biol. 2020 Dec;27(12):1105-1114. doi: 10.1038/s41594-020-0506-5. Epub 2020 Sep 14. Nat Struct Mol Biol. 2020. PMID: 32929283 Free PMC article.
-
Mini review: form and function in the human interphase chromosome.Cytogenet Cell Genet. 2000;90(1-2):13-21. doi: 10.1159/000015654. Cytogenet Cell Genet. 2000. PMID: 11060439 Review.
Cited by
-
Formation of Chromosomal Domains by Loop Extrusion.Cell Rep. 2016 May 31;15(9):2038-49. doi: 10.1016/j.celrep.2016.04.085. Epub 2016 May 19. Cell Rep. 2016. PMID: 27210764 Free PMC article.
-
Ultra-long-range interactions between active regulatory elements.Genome Res. 2023 Aug;33(8):1269-1283. doi: 10.1101/gr.277567.122. Epub 2023 Jul 14. Genome Res. 2023. PMID: 37451823 Free PMC article.
-
A maximum-entropy model for predicting chromatin contacts.PLoS Comput Biol. 2018 Feb 5;14(2):e1005956. doi: 10.1371/journal.pcbi.1005956. eCollection 2018 Feb. PLoS Comput Biol. 2018. PMID: 29401453 Free PMC article.
-
Mechanistic modeling of chromatin folding to understand function.Nat Methods. 2020 Aug;17(8):767-775. doi: 10.1038/s41592-020-0852-6. Epub 2020 Jun 8. Nat Methods. 2020. PMID: 32514111 Review.
-
Polymer Physics Models Reveal Structural Folding Features of Single-Molecule Gene Chromatin Conformations.Int J Mol Sci. 2024 Sep 23;25(18):10215. doi: 10.3390/ijms251810215. Int J Mol Sci. 2024. PMID: 39337699 Free PMC article.
References
-
- Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
