

There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS
- PMID: 27059391
- PMCID: PMC5003651
- DOI: 10.1016/j.brainres.2016.04.004
There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS
Abstract
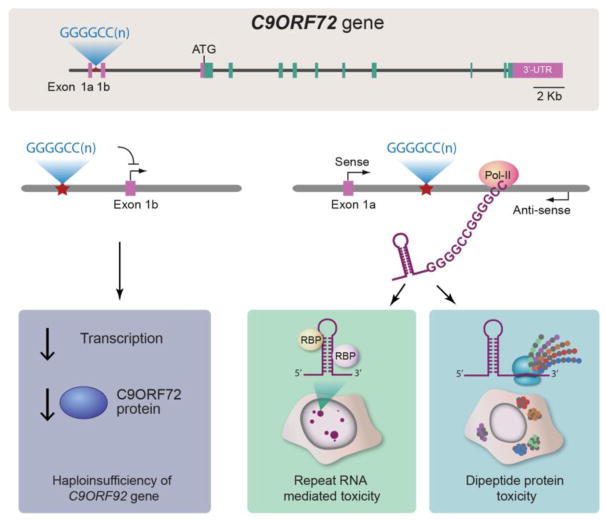
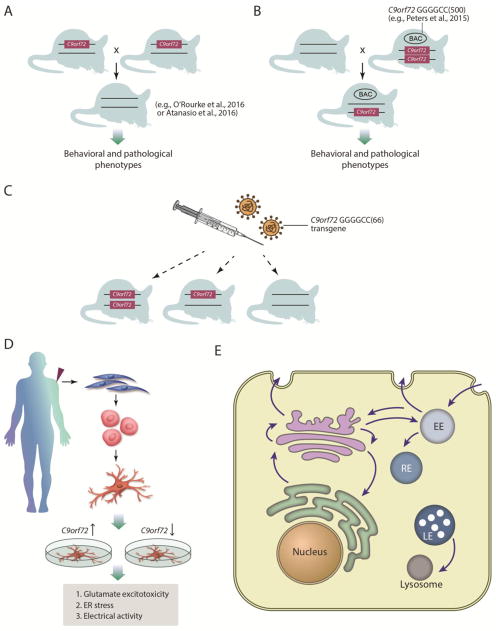
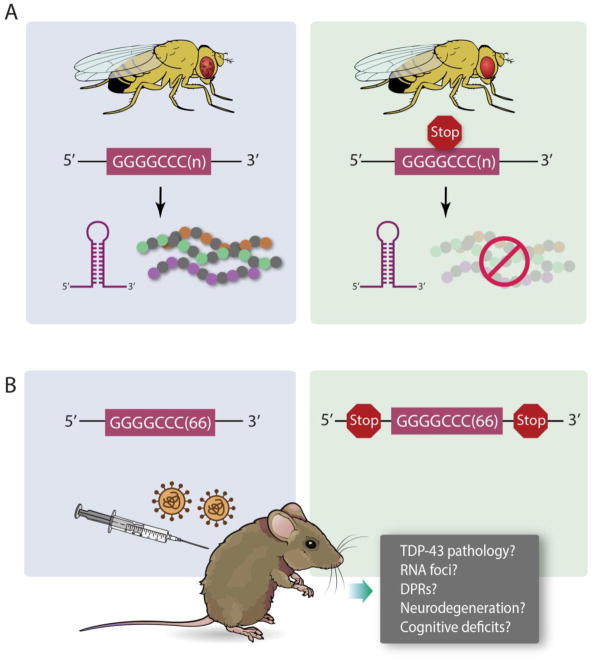
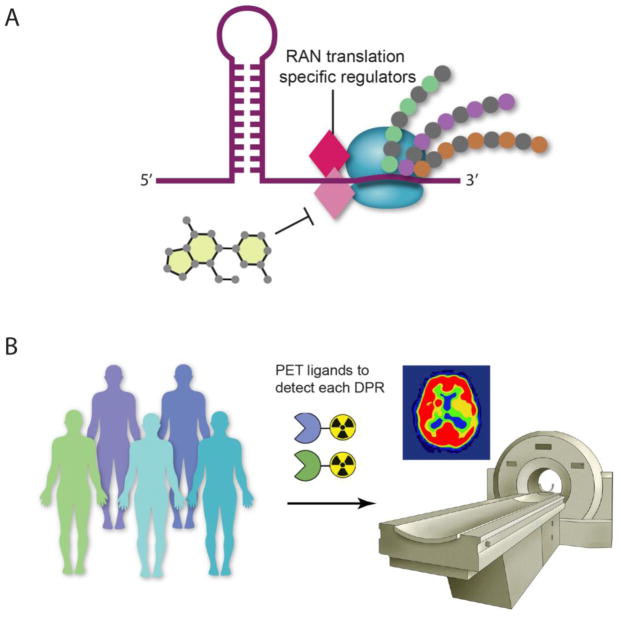
The discovery of C9orf72 mutations as the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) has awakened a surge of interest in deciphering how mutations in this mysterious gene cause disease and what can be done to stop it. C9orf72 harbors a hexanucleotide repeat, GGGGCC, in a non-coding region of the gene and a massive expansion of this repeat causes ALS, FTD, or both (FTD/ALS). Many questions lie ahead. What does this gene normally do? What is the consequence of an enormous GGGGCC repeat expansion on that gene's function? Could that hexanucleotide repeat expansion have additional pathological actions unrelated to C9orf72 function? There has been tremendous progress on all fronts in the quest to define how C9orf72 mutations cause disease. Many new experimental models have been constructed and unleashed in powerful genetic screens. Studies in mouse and human patient samples, including iPS-derived neurons, have provided unprecedented insights into pathogenic mechanisms. Three major hypotheses have emerged and are still being hotly debated in the field. These include (1) loss of function owing to decrease in the abundance of C9orf72 protein and its ability to carryout its still unknown cellular role; (2) RNA toxicity from bidirectionally transcribed sense (GGGGCC) and antisense (GGCCCC) transcripts that accumulate in RNA foci and might sequester critical RNA-binding proteins; (3) proteotoxicity from dipeptide repeat proteins produced by an unconventional form of translation from the expanded nucleotide repeats. Here we review the evidence in favor and against each of these three hypotheses. We also suggest additional experiments and considerations that we propose will help clarify which mechanism(s) are most important for driving disease and therefore most critical for considering during the development of therapeutic interventions. This article is part of a Special Issue entitled SI:RNA Metabolism in Disease.
Keywords: ALS, FTD; C9orf72; Dipeptide repeat protein; RNA.
Copyright © 2016 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Pathogenic determinants and mechanisms of ALS/FTD linked to hexanucleotide repeat expansions in the C9orf72 gene.Neurosci Lett. 2017 Jan 1;636:16-26. doi: 10.1016/j.neulet.2016.09.007. Epub 2016 Sep 13. Neurosci Lett. 2017. PMID: 27619540 Free PMC article. Review.
-
Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions.J Neurochem. 2016 Aug;138 Suppl 1:145-62. doi: 10.1111/jnc.13623. Epub 2016 Jun 15. J Neurochem. 2016. PMID: 27016280 Review.
-
Molecular Mechanisms of Neurodegeneration Related to C9orf72 Hexanucleotide Repeat Expansion.Behav Neurol. 2019 Jan 15;2019:2909168. doi: 10.1155/2019/2909168. eCollection 2019. Behav Neurol. 2019. PMID: 30774737 Free PMC article. Review.
-
Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice.Neuron. 2015 Dec 2;88(5):902-909. doi: 10.1016/j.neuron.2015.11.018. Neuron. 2015. PMID: 26637797 Free PMC article.
-
Therapeutic reduction of GGGGCC repeat RNA levels by hnRNPA3 suppresses neurodegeneration in Drosophila models of C9orf72-linked ALS/FTD.Hum Mol Genet. 2023 May 5;32(10):1673-1682. doi: 10.1093/hmg/ddac298. Hum Mol Genet. 2023. PMID: 36611007
Cited by
-
Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD.Mol Neurodegener. 2020 Jun 8;15(1):34. doi: 10.1186/s13024-020-00383-7. Mol Neurodegener. 2020. PMID: 32513219 Free PMC article. Review.
-
Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis.Sci Transl Med. 2017 Mar 29;9(383):eaai7866. doi: 10.1126/scitranslmed.aai7866. Sci Transl Med. 2017. PMID: 28356511 Free PMC article.
-
Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications.Lancet Neurol. 2018 Jan;17(1):94-102. doi: 10.1016/S1474-4422(17)30401-5. Epub 2017 Nov 16. Lancet Neurol. 2018. PMID: 29154141 Free PMC article. Review.
-
Mitophagy regulation in aging and neurodegenerative disease.Biophys Rev. 2023 Apr 4;15(2):239-255. doi: 10.1007/s12551-023-01057-6. eCollection 2023 Apr. Biophys Rev. 2023. PMID: 37124925 Free PMC article. Review.
-
Role of C9orf72 hexanucleotide repeat expansions in ALS/FTD pathogenesis.Front Mol Neurosci. 2024 Jan 22;17:1322720. doi: 10.3389/fnmol.2024.1322720. eCollection 2024. Front Mol Neurosci. 2024. PMID: 38318532 Free PMC article. Review.
References
-
- Aguzzi A, Gitler AD. A template for new drugs against Alzheimer’s disease. Cell. 2013;154:1182–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
