

MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification
- PMID: 26832969
- PMCID: PMC4740400
- DOI: 10.1038/ncomms10492
MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification
Abstract
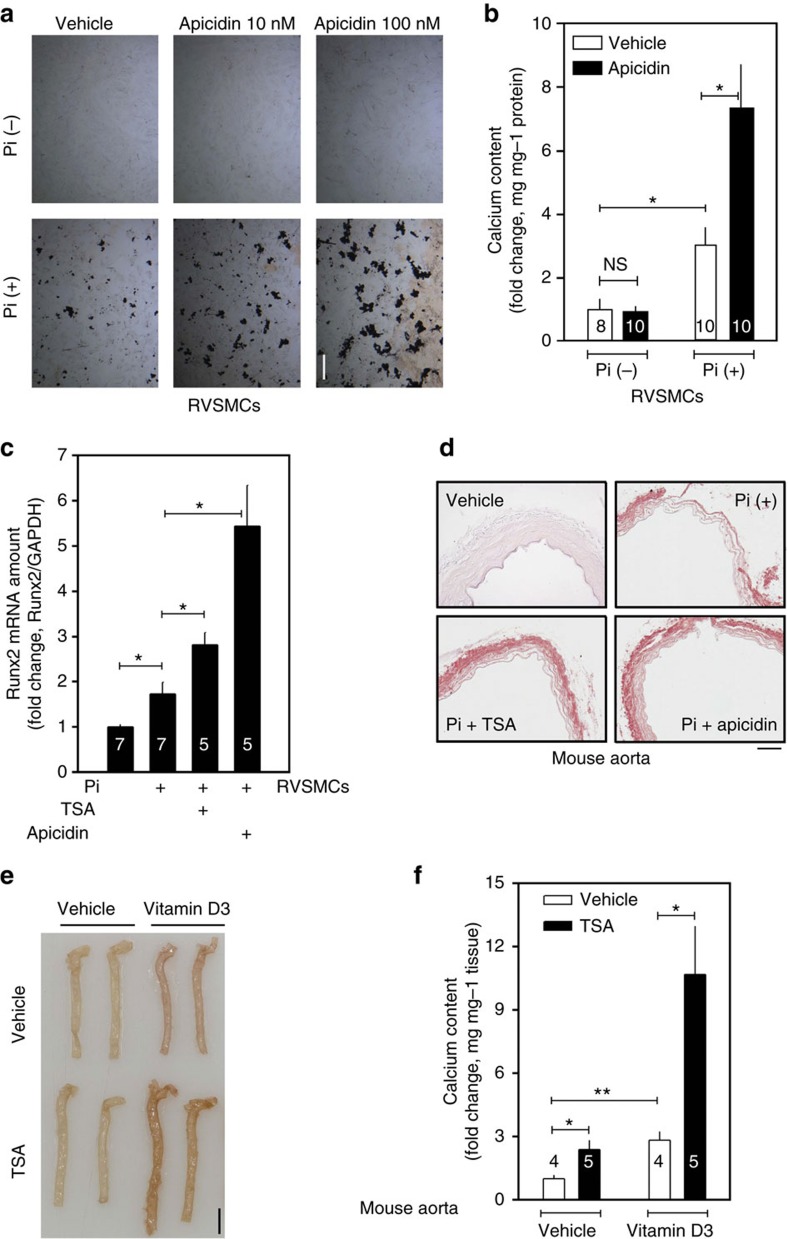
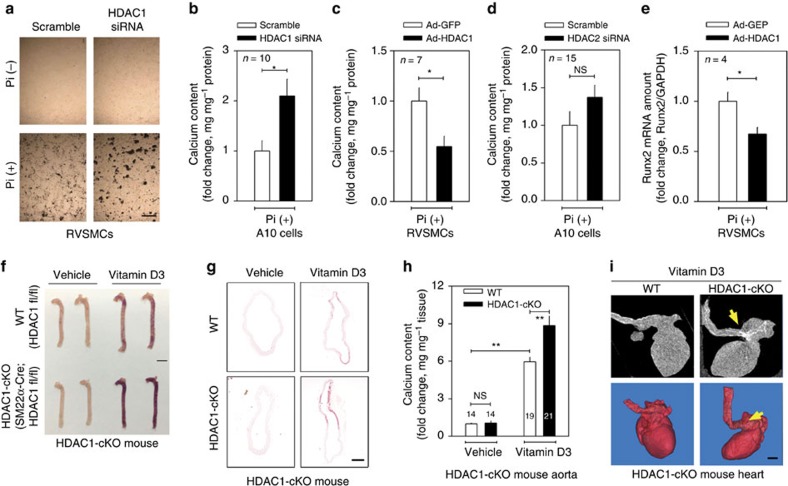
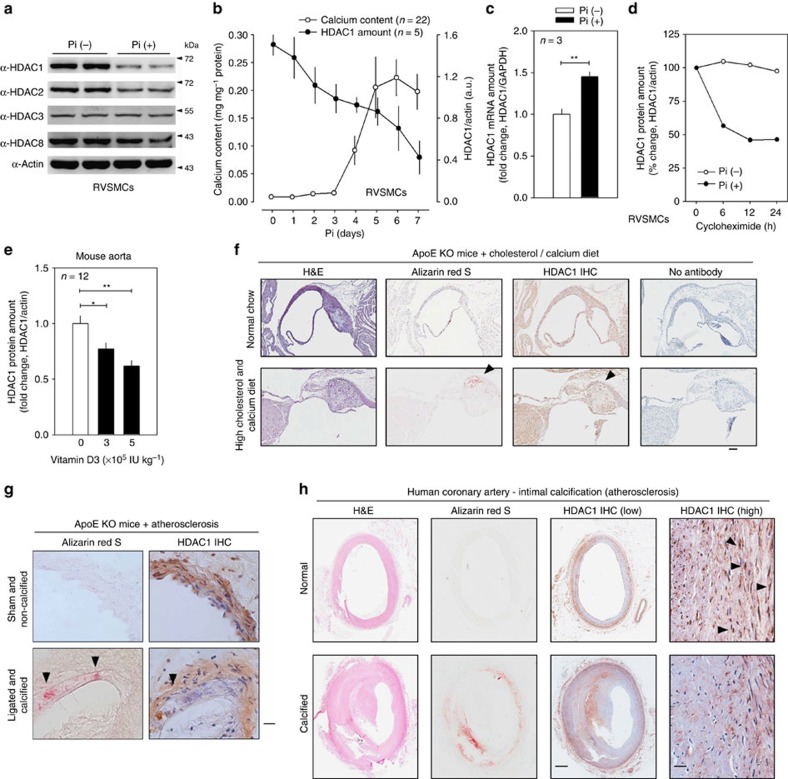
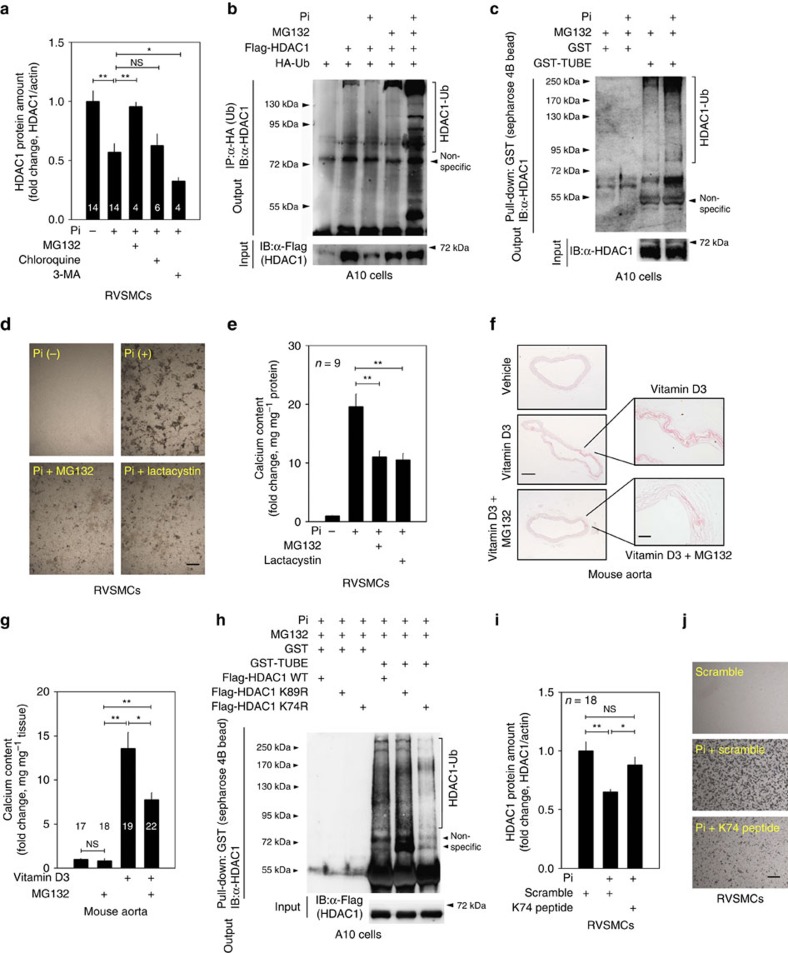
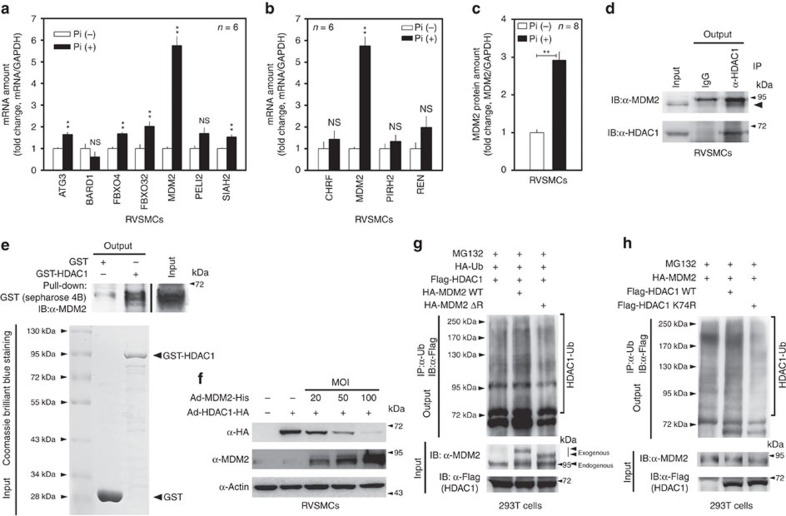
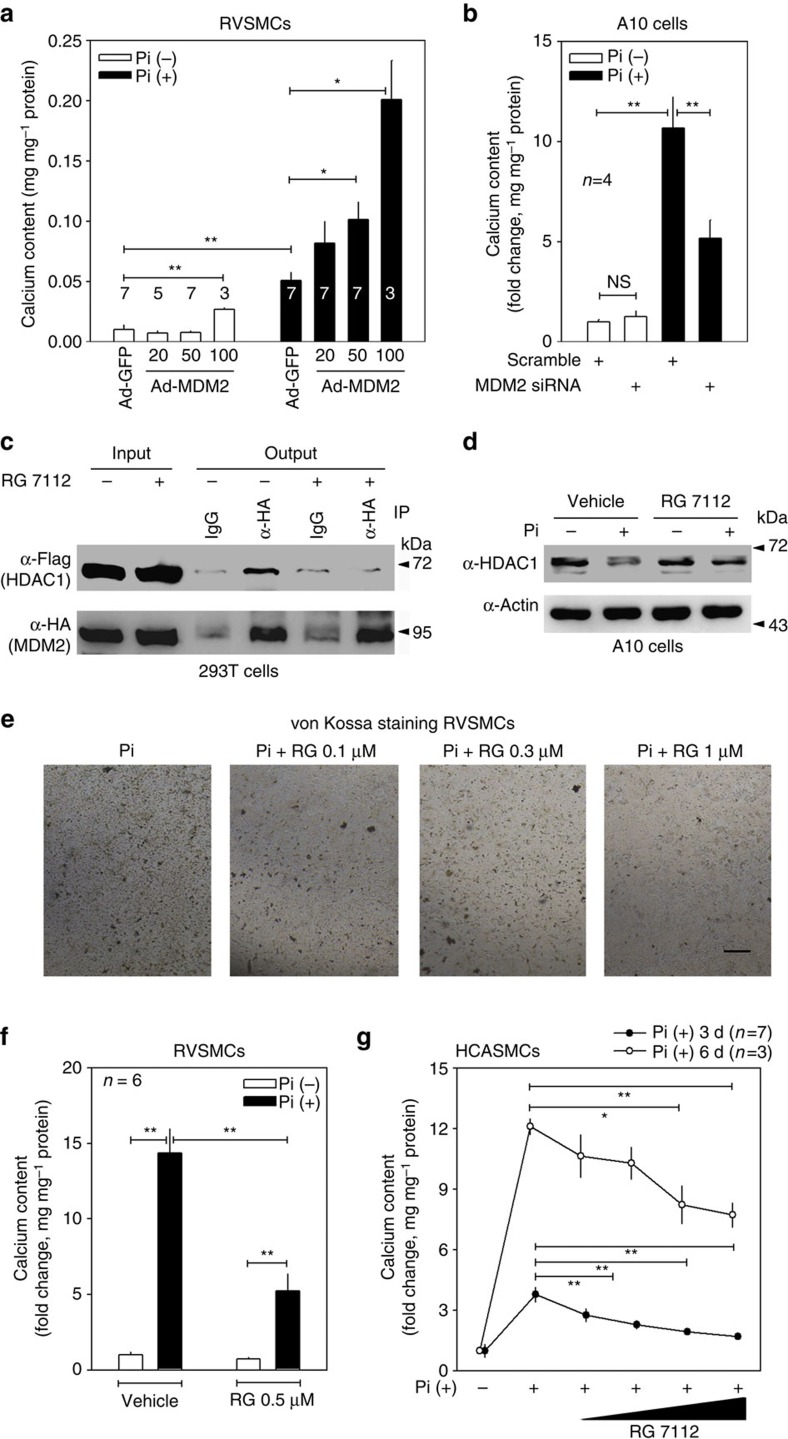
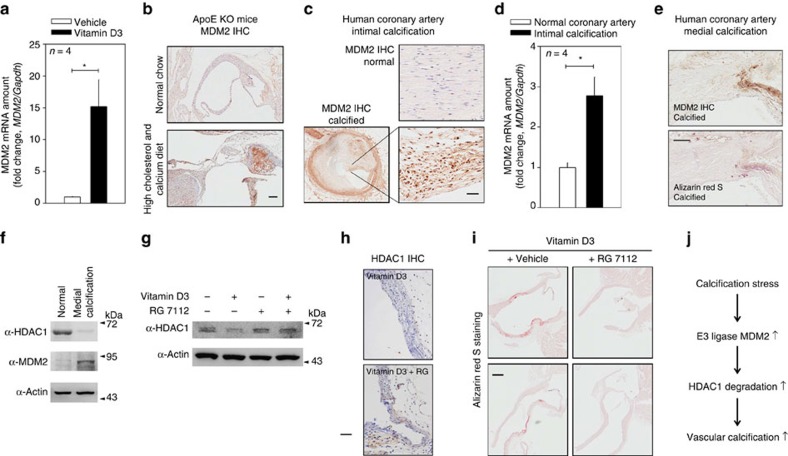
Vascular calcification (VC) is often associated with cardiovascular and metabolic diseases. However, the molecular mechanisms linking VC to these diseases have yet to be elucidated. Here we report that MDM2-induced ubiquitination of histone deacetylase 1 (HDAC1) mediates VC. Loss of HDAC1 activity via either chemical inhibitor or genetic ablation enhances VC. HDAC1 protein, but not mRNA, is reduced in cell and animal calcification models and in human calcified coronary artery. Under calcification-inducing conditions, proteasomal degradation of HDAC1 precedes VC and it is mediated by MDM2 E3 ubiquitin ligase that initiates HDAC1 K74 ubiquitination. Overexpression of MDM2 enhances VC, whereas loss of MDM2 blunts it. Decoy peptide spanning HDAC1 K74 and RG 7112, an MDM2 inhibitor, prevent VC in vivo and in vitro. These results uncover a previously unappreciated ubiquitination pathway and suggest MDM2-mediated HDAC1 ubiquitination as a new therapeutic target in VC.
Figures
Similar articles
-
HDAC1-mediated deacetylation of LSD1 regulates vascular calcification by promoting autophagy in chronic renal failure.J Cell Mol Med. 2020 Aug;24(15):8636-8649. doi: 10.1111/jcmm.15494. Epub 2020 Jun 28. J Cell Mol Med. 2020. PMID: 32596952 Free PMC article.
-
MDM2 accelerated renal senescence via ubiquitination and degradation of HDAC1.Acta Pharmacol Sin. 2024 Nov;45(11):2328-2338. doi: 10.1038/s41401-024-01294-9. Epub 2024 May 17. Acta Pharmacol Sin. 2024. PMID: 38760541
-
Regulation of MDM2 E3 ligase-dependent vascular calcification by MSX1/2.Exp Mol Med. 2021 Nov;53(11):1781-1791. doi: 10.1038/s12276-021-00708-6. Epub 2021 Nov 29. Exp Mol Med. 2021. PMID: 34845330 Free PMC article.
-
New Aspects of Vascular Calcification: Histone Deacetylases and Beyond.J Korean Med Sci. 2017 Nov;32(11):1738-1748. doi: 10.3346/jkms.2017.32.11.1738. J Korean Med Sci. 2017. PMID: 28960024 Free PMC article. Review.
-
p53 ubiquitination: Mdm2 and beyond.Mol Cell. 2006 Feb 3;21(3):307-15. doi: 10.1016/j.molcel.2006.01.020. Mol Cell. 2006. PMID: 16455486 Free PMC article. Review.
Cited by
-
Long noncoding RNAs in vascular smooth muscle cells regulate vascular calcification.Sci Rep. 2019 Apr 10;9(1):5848. doi: 10.1038/s41598-019-42283-x. Sci Rep. 2019. PMID: 30971745 Free PMC article.
-
Discovery of deoxyandrographolide and its novel effect on vascular senescence by targeting HDAC1.MedComm (2020). 2023 Aug 17;4(5):e338. doi: 10.1002/mco2.338. eCollection 2023 Oct. MedComm (2020). 2023. PMID: 37600507 Free PMC article.
-
Identification of XBP1-u as a novel regulator of the MDM2/p53 axis using an shRNA library.Sci Adv. 2017 Oct 18;3(10):e1701383. doi: 10.1126/sciadv.1701383. eCollection 2017 Oct. Sci Adv. 2017. PMID: 29057323 Free PMC article.
-
Epigenome alterations in aortic valve stenosis and its related left ventricular hypertrophy.Clin Epigenetics. 2017 Oct 3;9:106. doi: 10.1186/s13148-017-0406-7. eCollection 2017. Clin Epigenetics. 2017. PMID: 29026447 Free PMC article. Review.
-
OGT-Mediated KEAP1 Glycosylation Accelerates NRF2 Degradation Leading to High Phosphate-Induced Vascular Calcification in Chronic Kidney Disease.Front Physiol. 2020 Oct 26;11:1092. doi: 10.3389/fphys.2020.01092. eCollection 2020. Front Physiol. 2020. Retraction in: Front Physiol. 2024 Oct 09;15:1505634. doi: 10.3389/fphys.2024.1505634. PMID: 33192538 Free PMC article. Retracted.
References
-
- Frink R. J., Achor R. W., Brown A. L. Jr, Kincaid O. W. & Brandenburg R. O. Significance of calcification of the coronary arteries. Am. J. Cardiol. 26, 241–247 (1970). - PubMed
-
- Fitzgerald P. J., Ports T. A. & Yock P. G. Contribution of localized calcium deposits to dissection after angioplasty. An observational study using intravascular ultrasound. Circulation 86, 64–70 (1992). - PubMed
-
- Luo G. et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386, 78–81 (1997). - PubMed
-
- Kuro-o M. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 (1997). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
