

Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton
- PMID: 26824656
- PMCID: PMC4898774
- DOI: 10.1016/j.cell.2015.12.042
Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton
Abstract
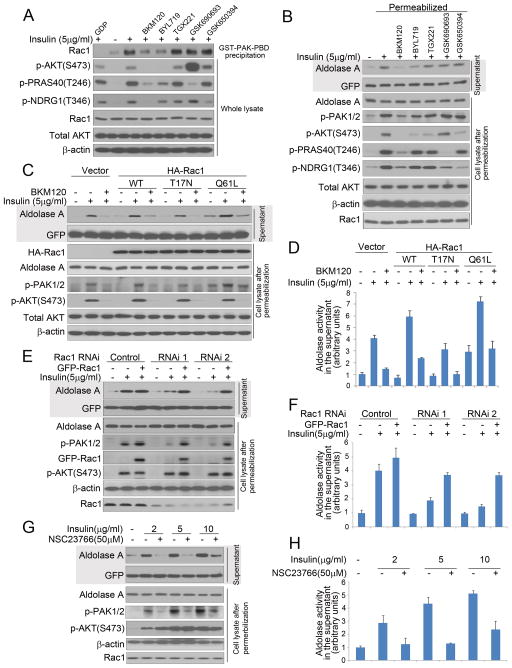
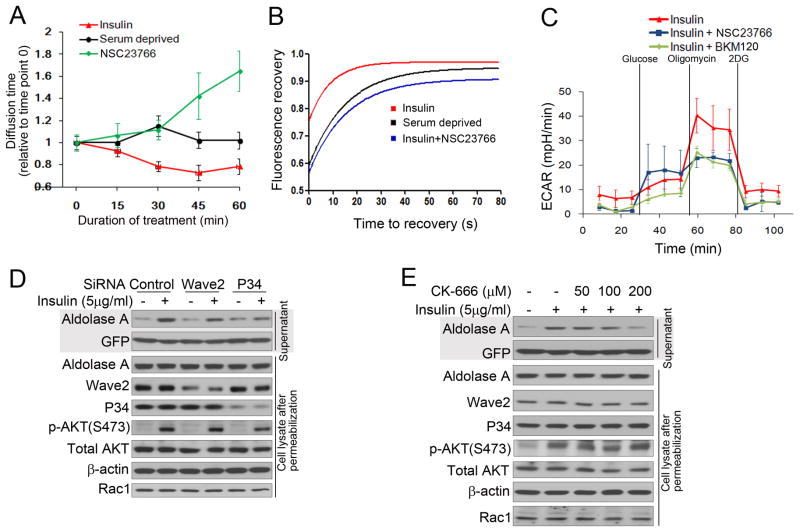
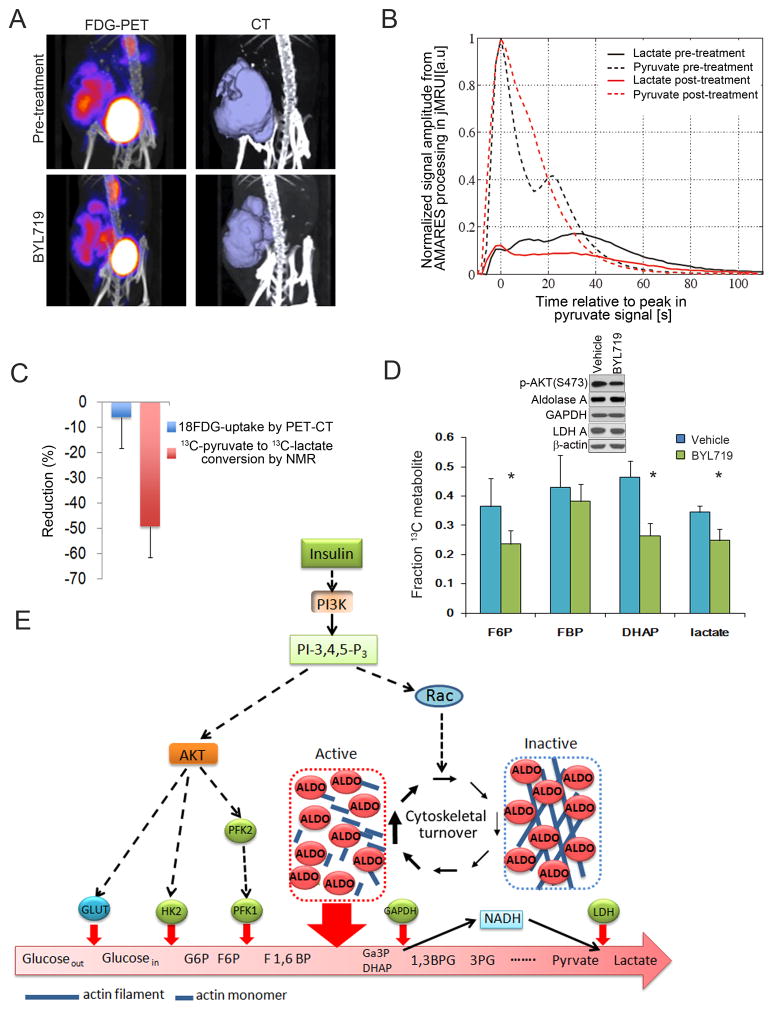
The phosphoinositide 3-kinase (PI3K) pathway regulates multiple steps in glucose metabolism and also cytoskeletal functions, such as cell movement and attachment. Here, we show that PI3K directly coordinates glycolysis with cytoskeletal dynamics in an AKT-independent manner. Growth factors or insulin stimulate the PI3K-dependent activation of Rac, leading to disruption of the actin cytoskeleton, release of filamentous actin-bound aldolase A, and an increase in aldolase activity. Consistently, PI3K inhibitors, but not AKT, SGK, or mTOR inhibitors, cause a significant decrease in glycolysis at the step catalyzed by aldolase, while activating PIK3CA mutations have the opposite effect. These results point toward a master regulatory function of PI3K that integrates an epithelial cell's metabolism and its form, shape, and function, coordinating glycolysis with the energy-intensive dynamics of actin remodeling.
Copyright © 2016 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
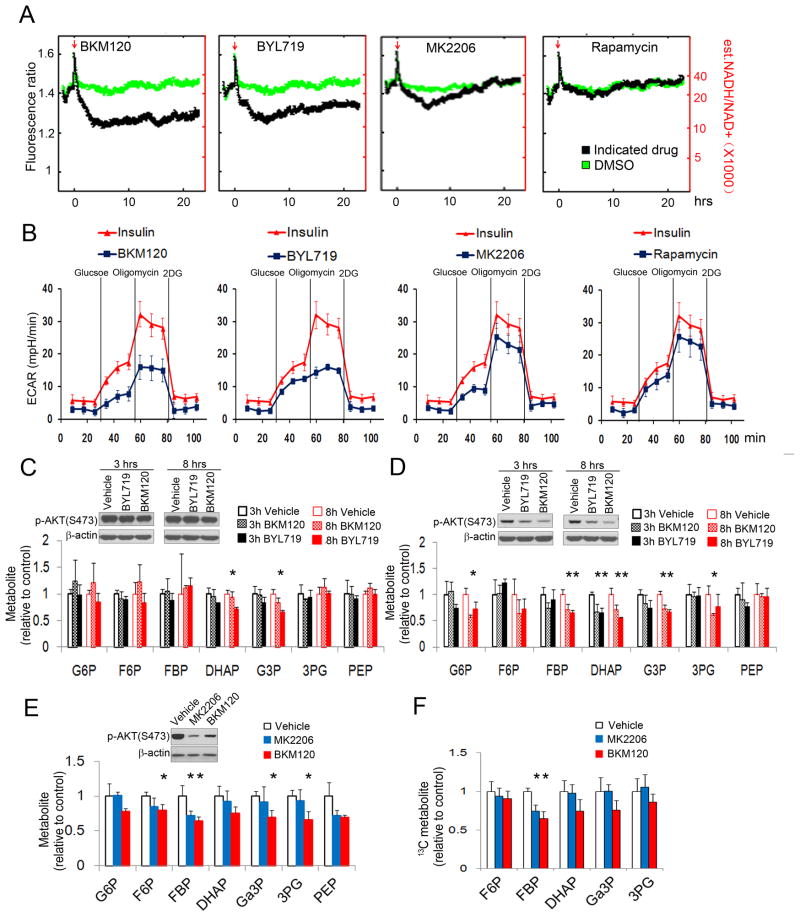
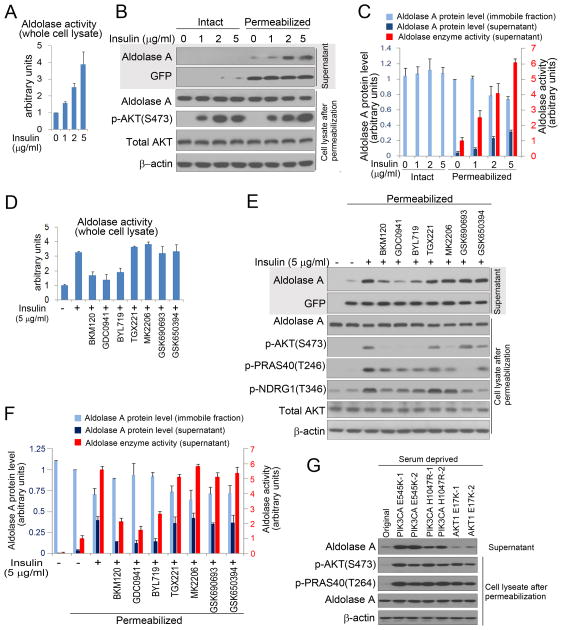
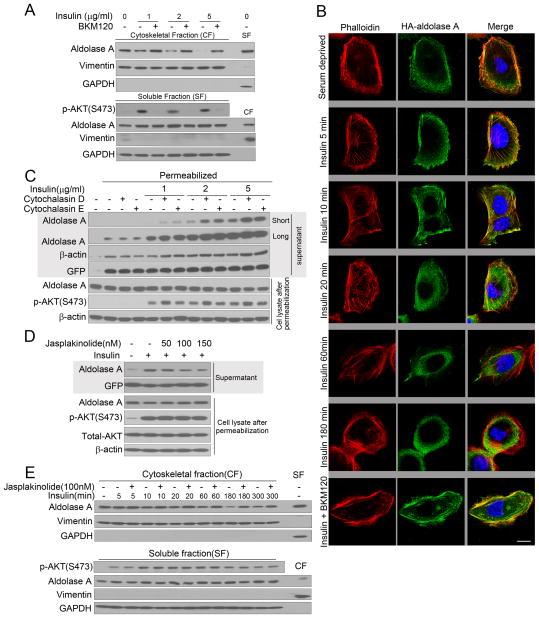
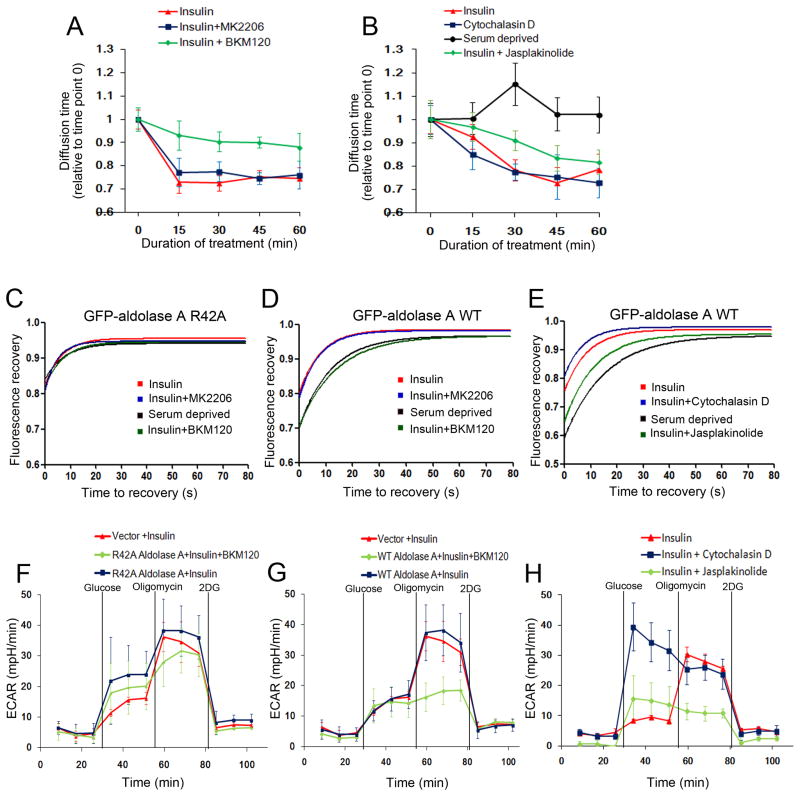
Similar articles
-
Reconstitution of the mammalian PI3K/PTEN/Akt pathway in yeast.Biochem J. 2005 Sep 1;390(Pt 2):613-23. doi: 10.1042/BJ20050574. Biochem J. 2005. PMID: 15913452 Free PMC article.
-
In guinea pig sperm, aldolase A forms a complex with actin, WAS, and Arp2/3 that plays a role in actin polymerization.Reproduction. 2009 Apr;137(4):669-78. doi: 10.1530/REP-08-0353. Epub 2009 Jan 16. Reproduction. 2009. PMID: 19151127
-
Effects of orexin A on glucose metabolism in human hepatocellular carcinoma in vitro via PI3K/Akt/mTOR-dependent and -independent mechanism.Mol Cell Endocrinol. 2016 Jan 15;420:208-16. doi: 10.1016/j.mce.2015.11.002. Epub 2015 Nov 6. Mol Cell Endocrinol. 2016. PMID: 26549689
-
Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer.Oncologist. 2011;16 Suppl 1:12-9. doi: 10.1634/theoncologist.2011-S1-12. Oncologist. 2011. PMID: 21278436 Review.
-
Targeting phosphoinositide 3-kinase: moving towards therapy.Biochim Biophys Acta. 2008 Jan;1784(1):159-85. doi: 10.1016/j.bbapap.2007.10.003. Epub 2007 Oct 12. Biochim Biophys Acta. 2008. PMID: 17997386 Review.
Cited by
-
Extracellular Matrix Stiffness: New Areas Affecting Cell Metabolism.Front Oncol. 2021 Feb 24;11:631991. doi: 10.3389/fonc.2021.631991. eCollection 2021. Front Oncol. 2021. PMID: 33718214 Free PMC article. Review.
-
Reciprocal regulation of cellular mechanics and metabolism.Nat Metab. 2021 Apr;3(4):456-468. doi: 10.1038/s42255-021-00384-w. Epub 2021 Apr 19. Nat Metab. 2021. PMID: 33875882 Free PMC article. Review.
-
Aldolase A promotes cell proliferation and cisplatin resistance via the EGFR pathway in gastric cancer.Am J Transl Res. 2022 Sep 15;14(9):6586-6595. eCollection 2022. Am J Transl Res. 2022. PMID: 36247245 Free PMC article.
-
PI3K/Akt pathway-mediated HO-1 induction regulates mitochondrial quality control and attenuates endotoxin-induced acute lung injury.Lab Invest. 2019 Dec;99(12):1795-1809. doi: 10.1038/s41374-019-0286-x. Epub 2019 Sep 30. Lab Invest. 2019. PMID: 31570770
-
Spatial Regulation of Mitochondrial Heterogeneity by Stromal Confinement in Micropatterned Tumor Models.Sci Rep. 2019 Aug 1;9(1):11187. doi: 10.1038/s41598-019-47593-8. Sci Rep. 2019. PMID: 31371796 Free PMC article.
References
-
- Albe KR, Butler MH, Wright BE. Cellular concentrations of enzymes and their substrates. J Theor Biol. 1990;143:163–195. - PubMed
-
- Arnold H, Pette D. Binding of aldolase and triosephosphate dehydrogenase to F-actin and modification of catalytic properties of aldolase. European journal of biochemistry/FEBS. 1970;15:360–366. - PubMed
-
- Barthel A, Kohn AD, Luo Y, Roth RA. A constitutively active version of the Ser/Thr kinase Akt induces production of the ob gene product, leptin, in 3T3-L1 adipocytes. Endocrinology. 1997;138:3559–3562. - PubMed
-
- Benard V, Bohl BP, Bokoch GM. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. The Journal of biological chemistry. 1999;274:13198–13204. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R21 EB014471/EB/NIBIB NIH HHS/United States
- 5P01CA120964-05/CA/NCI NIH HHS/United States
- R01 CA169470/CA/NCI NIH HHS/United States
- P30 CA046592/CA/NCI NIH HHS/United States
- P01 CA120964/CA/NCI NIH HHS/United States
- R01 CA152330/CA/NCI NIH HHS/United States
- R37 GM041890/GM/NIGMS NIH HHS/United States
- R01 GM041890/GM/NIGMS NIH HHS/United States
- 5P30CA006516-46/CA/NCI NIH HHS/United States
- R01CA152330-0/CA/NCI NIH HHS/United States
- GM041890/GM/NIGMS NIH HHS/United States
- S10 OD010612/OD/NIH HHS/United States
- P30 CA006516/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
